
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
Exploiting Metabolic Reprogramming and Its Therapeutic Vulnerabilities in Prostate Cancer
Ukasha Ahmed1
1Department of Biochemistry, School of Medicine, University of Texas at Austin, 110 Inner Campus Dr, TX 78712, Austin, Texas State, United States.
Correspondence: Ukasha Ahmed (Department of Biochemistry, School of Medicine, University of Texas at Austin, 110 Inner Campus Dr, TX 78712, Austin, Texas State, United States; Email: Ukasha602@gmail.com).
Annals of Urologic Oncology 2025, 8(3): 127-137. https://doi.org/10.32948/auo.2025.11.09
Received: 09 Sep 2025 | Accepted: 29 Oct 2025 | Published online: 18 Nov 2025
Prostate cancer is a metabolically distinct malignancy as it exhibits strong flexibility in how it uses energy and perform physiological processes. In contrast to many solid tumors which mostly depend on aerobic glycolysis, the primary prostate cancer cells still depend mainly on oxidative phosphorylation and tricarboxylic acid (TCA) cycle for energy supply. This unique metabolic pattern is mainly controlled by androgen receptor signaling and also affected by mitochondrial functions and zinc level inside the cells. When the disease goes into advanced stage, especially castration-resistant prostate cancer (CRPC), the tumor cells change their energy system toward glycolytic and lipogenic ways to support hyperactive cell cycle, therapy resistance, and metastasis. This review gives a detailed discussion about metabolic reprogramming in prostate cancer with focus on glycolysis, mitochondrial dysfunction, and dysregulated lipid and cholesterol metabolism. Key enzymes, transporters and transcription factors, such as GLUT1, HK2, PFK1, PKM2, LDHA, PDK, FABP5, ACLY, ACAC, FASN, SREBPs and LXRs are discussed as important players of tumor bioenergetics , and as possible drug targets. Especially lipid metabolism has shown strong relation with CRPC aggressiveness, which is promoted by androgen receptor-controlled increase of lipogenic enzymes and fatty acid transport process. Interaction between metabolic pathways and oncogenic signaling like PI3K/AKT/mTOR makes the situation more complex. The review also covers new therapeutic strategies which make use of these metabolic vulnerabilities, including small molecule inhibitors, natural substances, and combination treatments. Better understanding of metabolic reprogramming of prostate cancer at different disease stages can help in creating more specific therapies to overcome resistance and improve clinical outcomes.
Key words prostate cancer, metabolic reprogramming, mitochondrial dysfunction, glycolysis, oxidative phosphorylation, lipid metabolism, targeted therapy
Metabolic reprogramming is now considered as a hallmark of cancer and helps tumor cells to adjust under changing nutrients and oxygen levels [10]. In prostate cancer, this metabolic change involves glucose, lipid, and amino acid metabolism. While most cancers rely mainly on aerobic glycolysis (Warburg effect), the early stage prostate cancer keeps oxidative phosphorylation and tricarboxylic acid (TCA) cycle as main energy sources, which is strongly controlled by androgen receptor signaling [11]. When disease advances to CRPC, metabolism shifts more toward glycolysis and lipid biosynthesis, which support tumor survival, growth, and therapy resistance [12]. Among these metabolic processes, lipid metabolism plays a key role. The disturbed de novo lipogenesis, fatty acid uptake, and β-oxidation give cells both structural and signaling components that drive tumor progression [13, 14]. androgen receptor signaling also controls enzymes responsible for steroidogenesis and fatty acid oxidation, thus helping cancer cell survival even in low androgen condition [15]. High lipid accumulation, especially of glycerophospholipids, is linked with therapy resistant CRPC [16]. This combined dependency on oxidative phosphorylation and lipid metabolism makes prostate cancer different from many other solid tumors and provides a group of new therapeutic targets.
This review mainly discusses the metabolic reprogramming in prostate cancer and how glycolysis, mitochondrial function, and lipid metabolism work together. It also describes new metabolic targets and therapeutic strategies which may help in improving the outcome in both hormone-sensitive and castration-resistant stages.
Prostate cancer also shows high de novo fatty acid synthesis which is controlled by key enzymes such as ATP citrate lyase (ACLY), acetyl-CoA carboxylase (ACAC), and fatty acid synthase (FASN). ACLY converts citrate into acetyl-CoA, then ACAC changes it into malonyl-CoA, and FASN uses malonyl-CoA to make palmitate. Palmitate is further modified by stearoyl-CoA desaturase and ELOVL enzymes to form complex lipids like triacylglycerols (Figure 3) [29, 30]. These lipogenic enzymes are often increased in prostate cancer and are also related to androgen receptor and PI3K/AKT/mTOR signaling that promote lipid production and storage [31]. As a result, there is higher amount of phospholipids, sphingolipids, and triglycerides accumulated in lipid droplets, which is known as “lipogenic phenotype.” This type of metabolism is more common in metastatic CRPC and shows aggressive behavior [32]. Besides new lipid formation, prostate cancer cells also increase fatty acid uptake and transport using membrane proteins such as CD36 (fatty acid translocase), fatty acid transport proteins (FATPs), and fatty acid-binding proteins (FABPs) [33]. In PTEN-deficient models, deleting CD36 reduces fatty acid uptake, decreases oncogenic lipid content, and slows tumor progression [34]. CD36 is also related to metastasis because metastatic cells need fatty acid uptake for colonizing new sites [35]. High CD36 expression is connected with poor prognosis [36]. FATPs, especially FATP6, are found in high amount in prostate cancer and linked with lower survival [37]. FABPs like FABP4 and FABP5 help in moving fatty acids inside the cell. FABP4 interacts with peroxisome proliferator-activated receptor gamma (PPARγ), thereby helping in proliferation and differentiation [38]. FABP5 is hyperactive in advanced prostate cancer cases, where it supports tumor growth by sending fatty acids to PPARγ, which, in turn, activates genes like vascular endothelial growth factor (VEGF) [39]. FABP5 can also regulate the expression of AR-V7, keeping CRPC growing even under androgen receptor targeted therapy [40].
Fatty acid β-oxidation (FAO) presents another source of energy in prostate cancer cells (Figure 2), especially under nutrient-deprived conditions. Carnitine palmitoyltransferase 1A (CPT-1A) is the main enzyme that controls FAO and is found high in prostate cancer. Inhibition of CPT-1A, together with androgen receptor blockers like enzalutamide, can reduce tumor growth by changing AKT activity and activating the INPP5K pathway [41]. CPT-1B, another isoform controlled by androgen receptor, also helps in castration resistance by maintaining AKT signaling [42]. α-Methylacyl-CoA racemase (AMACR), a peroxisomal enzyme involved in β-oxidation of branched fatty acids, is strongly expressed in prostate cancer and is used as diagnostic and therapeutic marker [43]. Interestingly, in neuroendocrine prostate cancer, which is a very aggressive type, tumor cells depend less on FAO and more on glutamine metabolism. They show low kidney-type glutaminase and high glutaminase 1 (GLS1) xpression to adjust under nutrient stress and therapy [3]. Prostate cancer also increases cholesterol biosynthesis through the mevalonate pathway. Cholesterol works as a membrane part and also as precursor for androgen synthesis [44]. The enzyme 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) is the main step in cholesterol production and is high in enzalutamide-resistant cells, connecting cholesterol metabolism with drug resistance [45]. Statins, which block HMGCR, can reduce this resistance and slow tumor growth [46]. Prostate cancer cells also improve cholesterol uptake by increasing low-density lipoprotein (LDL) receptors and changing ATP-binding cassette (ABC) transporters [32]. High cholesteryl ester level in lipid droplets, promoted by sterol regulatory element-binding protein 2 (SREBP2) and LDL receptors, supports aggressive behavior [47]. Loss of PTEN, which is common in prostate cancer, activates PI3K/AKT/mTOR pathway that promotes cholesterol storage and helps cell survival [32]. In general, prostate cancer metabolism shows dynamic changes in mitochondrial respiration, glycolysis, lipid production, fatty acid oxidation, and cholesterol metabolism (Figure 1-3). These pathways are tightly controlled by oncogenic signals and change under therapy stress. Understanding their interaction can help in designing combination treatments to target these metabolic weaknesses and overcome drug resistance in prostate cancer.
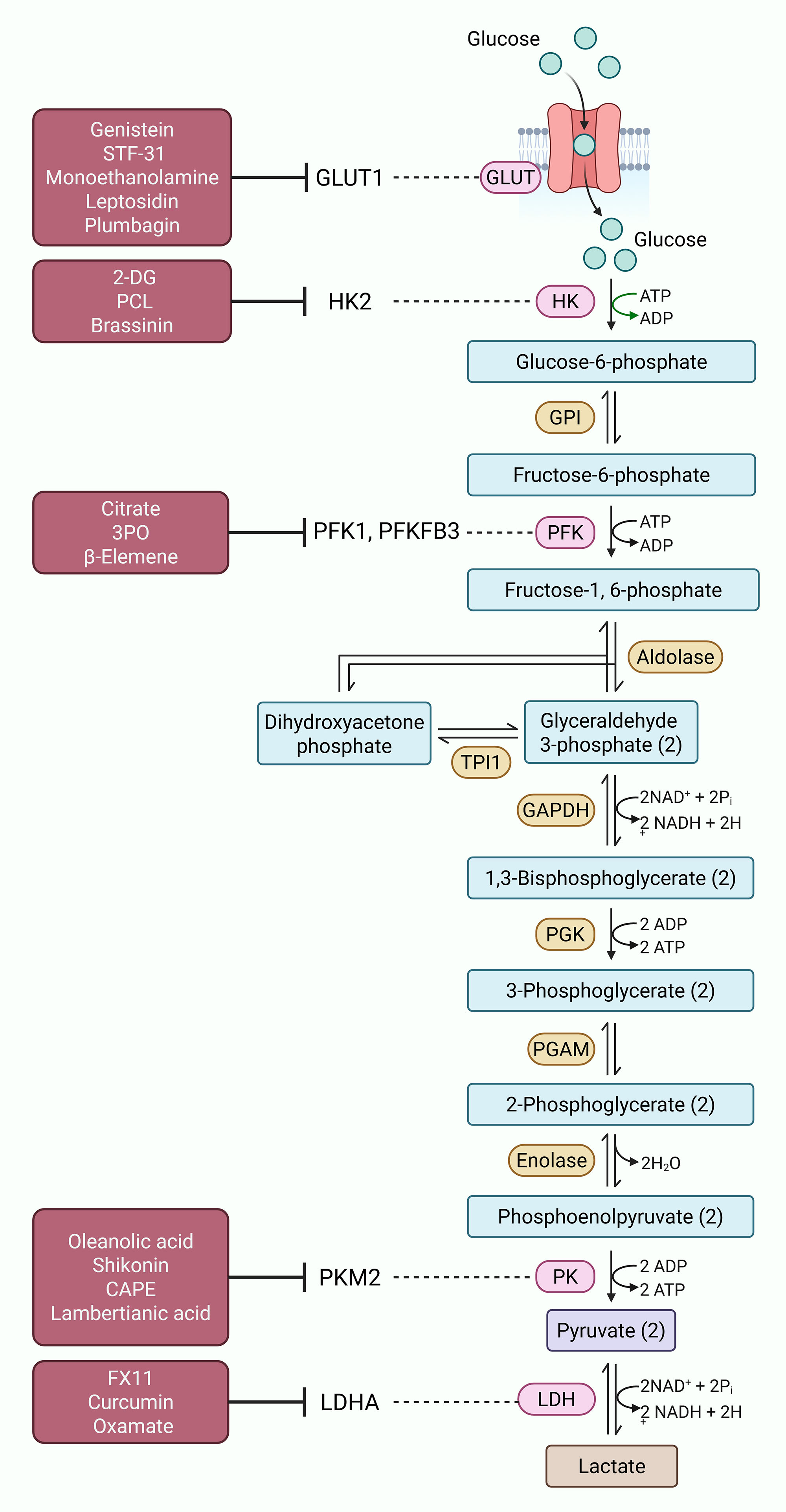 Figure 1. Targeting glycolysis in prostate cancer. Cancer cells often rely on glycolysis for energy production. Various enzymes involved in this process, including GLUT1, HK2, PFK1, PFKFB3, PKM2 and LDHA, are often dysregulated in prostate cancer cells, fueling tumor progression and disease aggressiveness. Different inhibitors, both synthetic and natural (highlighted in maroon boxes), have shown potential to target these metabolic vulnerabilities of glycolysis and alleviate disease aggressiveness in prostate cancer.
Figure 1. Targeting glycolysis in prostate cancer. Cancer cells often rely on glycolysis for energy production. Various enzymes involved in this process, including GLUT1, HK2, PFK1, PFKFB3, PKM2 and LDHA, are often dysregulated in prostate cancer cells, fueling tumor progression and disease aggressiveness. Different inhibitors, both synthetic and natural (highlighted in maroon boxes), have shown potential to target these metabolic vulnerabilities of glycolysis and alleviate disease aggressiveness in prostate cancer.
Hexokinase 2 (HK2) catalyzes the first step of glycolysis and is found high in prostate cancer, mainly in low oxygen conditions. It helps the cells to survive through aerobic glycolysis [57]. 2-Deoxy-D-glucose (2-DG), a glucose analog, inhibits HK activity and lowers glycolytic rate. Early trials showed some benefit but long-term use caused resistance [58]. When 2-DG is combined with metformin, an autophagy inhibitor, apoptosis becomes stronger [59]. Polysaccharide C-type lectin (PCL), a mannose-specific lectin, interacts with epidermal growth factor receptor (EGFR) and lowers HK2 expression-driven glycolysis, ultimately promoting apoptosis [60]. Brassinin, a natural phytoalexin from cruciferous vegetables, blocks MAPK pathway, leading to HK2 inhibition, ROS production and cell death [61]. Phosphofructokinase-1 (PFK1) and its regulator PFKFB (6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase) control the speed of glycolysis. In prostate cancer, PFK1 and PFKFB3 are usually higher and increase glycolytic rate for tumor survival [62]. Citrate, which naturally inhibits PFK1, can reduce both glycolysis and TCA cycle, resulting in low energy and slow tumor growth [63]. Some small molecules such as 3PO block PFKFB3 by inhibiting fructose-2,6-bisphosphate synthesis, which reduces glucose consumption, increases ROS, and induces autophagy [64]. β-Elemene, a plant compound from traditional Chinese medicine, decreases PFKFB3 expression, reduces lactate formation, slows proliferation, and improves sensitivity to chemotherapy [65]. Thus, PFK and PFKFB3 are considered as important therapeutic targets for metabolic treatment of prostate cancer.
PKM2 controls the final step of glycolysis. Natural compounds like oleanolic acid reduce PKM2 level, induce apoptosis, and cell cycle arrest in prostate cancer cells [66]. Shikonin, a naphthoquinone compound, inhibits PKM2 and increases ROS together with AMPK activation. It is highly effective when used in combination with chemotherapeutic drugs like cabazitaxel [67, 68]. Caffeic acid phenethyl ester (CAPE) also decreases glycolysis and androgen receptor signaling, which causes toxicity in prostate cancer cells [69]. Lambertianic acid, obtained from Pinus species, reduces lactate production and inhibits PKM2 phosphorylation, disturbing PKM2/β-catenin axis that controls tumor growth [70]. LDH, mainly its LDHA isoform, plays a main role in converting pyruvate to lactate and maintaining Warburg effect. LDHA is overexpressed in prostate cancer and linked with tumor growth and drug resistance [71]. Different LDHA inhibitors show promising preclinical activity. FX11, a competitive LDHA inhibitor, reduces lactate generation, decreases glucose uptake, increases oxidative stress, and stops metastasis [72]. Curcumin, a polyphenolic compound from Curcuma longa, induces endoplasmic reticulum stress, increases ROS, and upregulates pro-apoptotic genes. It also reduces LDH expression and affects CD44 positive prostate cancer cell survival [73, 74]. Oxamate, a pyruvate analog, blocks LDH competitively and lowers lactate production. Combination of oxamate (or sodium oxamate) with docetaxel in CRPC models gives better results, reducing tumor growth and improving drug response [75]. These studies suggest that PKM2 and LDHA, are important metabolic targets and their inhibition could be useful, particularly in combination therapy approaches.
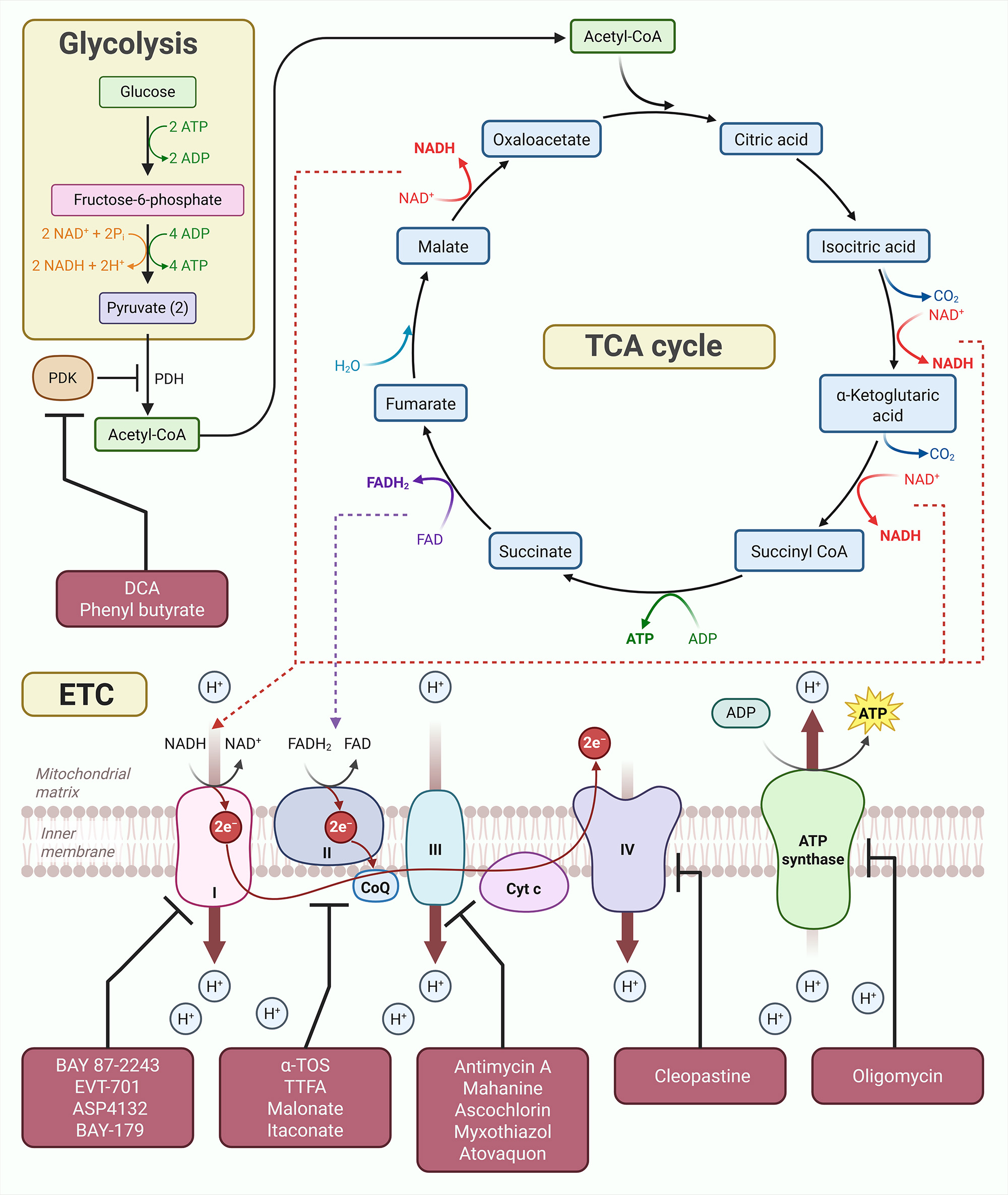 Figure 2. Targeting mitochondrial dysfunction in prostate cancer. As pyruvate exits glycolysis, it is converted into Acetyl-CoA, which serves as fuel for tricarboxylic acid (TCA) cycle. Co-enzymes, NADH and FADH2 produced in TCA cycle then aid electron transport chain complexes in the inter-membrane space of mitochondria to produce energy in the form of ATP. Various enzymes involved in different steps of this process are often dysregulated in prostate cancer cells, leading to mitochondrial dysfunction and tumor progression during early stages of prostate cancer. Different inhibitors, both synthetic and natural (highlighted in maroon boxes), have shown potential to target these metabolic vulnerabilities of mitochondrial dysfunction and alleviate disease aggressiveness in prostate cancer.
Figure 2. Targeting mitochondrial dysfunction in prostate cancer. As pyruvate exits glycolysis, it is converted into Acetyl-CoA, which serves as fuel for tricarboxylic acid (TCA) cycle. Co-enzymes, NADH and FADH2 produced in TCA cycle then aid electron transport chain complexes in the inter-membrane space of mitochondria to produce energy in the form of ATP. Various enzymes involved in different steps of this process are often dysregulated in prostate cancer cells, leading to mitochondrial dysfunction and tumor progression during early stages of prostate cancer. Different inhibitors, both synthetic and natural (highlighted in maroon boxes), have shown potential to target these metabolic vulnerabilities of mitochondrial dysfunction and alleviate disease aggressiveness in prostate cancer.
SREBP1 and SREBP2 act as main transcription regulators for genes involved in fatty acid and cholesterol production. Blocking SREBP activity has shown promise as a therapeutic approach. Fatostatin, a small-molecule inhibitor which stops SREBP from binding to SREBP cleavage-activating protein (SCAP), shows strong anticancer effect in prostate cancer models [100]. In cell line studies, fatostatin reduces proliferation, migration, and invasion, and in animal models it causes cell cycle arrest and apoptosis [101]. Fatostatin also increases the effect of docetaxel chemotherapy in both androgen receptor-positive and -negative prostate cancer cells. The combination effect is higher in cells with TP53 mutation, suggesting fatostatin may be helpful to overcome therapy resistance [102]. Studies in mouse models further show that fatostatin reduces tumor growth and lymph node metastasis [101]. Several natural products also show activity against lipid metabolism in prostate cancer. Extracts from Withania somnifera inhibit SREBP1, FASN and ACAC, thereby disturbing lipid synthesis and inducing apoptosis in cancer cells [103]. Eriobotrya japonica extract targets both lipid and androgen receptor signaling by blocking the SREBP1, resulting in diminished androgen receptor level and induction of apoptosis [104]. The medicinal fungus Ganoderma tsugae exert its anticancer effects by suppressing SREBP-driven lipogenesis as well [105]. Betulin, a plant-based triterpenoid, suppresses SREBP1 and reduces glutathione peroxidase 4 (GPX4) expression, resulting in ferroptosis induction [106]. Silibinin, a flavonolignan from milk thistle, prevents nuclear transport of SREBP1 through AMPK activation, reducing lipid and cholesterol storage and slowing androgen-independent prostate cancer growth [99]. Cholesterol metabolism is another main area for treatment. Statins, which inhibit 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCR), the key enzyme of the mevalonate pathway, reduce prostate cancer proliferation and migration by inducing apoptosis and cell cycle arrest [107]. Among statins, simvastatin shows strong tumor-suppressive effect in xenograft models [108]. Liver X receptors (LXRs) are also involved in lipid control and prostate cancer. Activation of LXRs can block epithelial-to-mesenchymal transition, which is a key step in metastasis [109]. The LXR-α agonist GW3965 activates tumor-suppressive signaling, while the EGFR inhibitor Afatinib raises LXR-α expression by blocking AKT and activating FOXO3A. Combination of Afatinib and GW3965 gives a synergistic antitumor effect [110]. In summary, lipid and cholesterol metabolism give many promising therapeutic targets in prostate cancer. Blocking FABPs, ACLY, ACAC, FASN, HMGCR, and SREBPs, and promoting LXRs (Figure 3) can disrupt energy homeostasis and results in good clinical outcomes, especially in advanced and resistant prostate cancer cases.
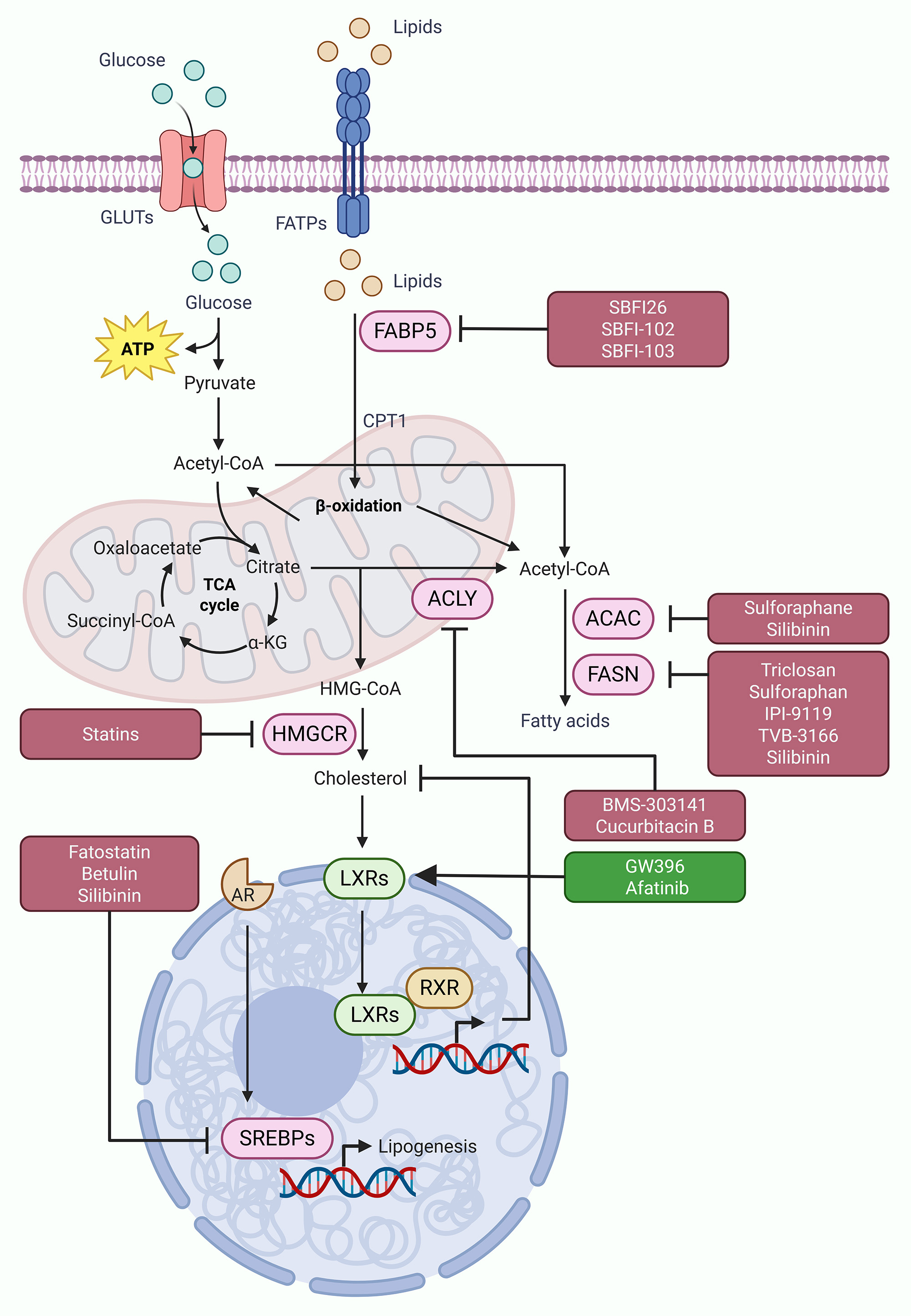 Figure 3. Targeting dysregulated lipid metabolism in prostate cancer. Dsyregulated fatty acid and cholesterol metabolism often contribute to tumor progression and disease aggressiveness in prostate cancer. Various enzymes and transcription factors involved in this process, including FABPs, ACLY, ACAC, FASN, HMGCR, and SREBPs, are often dysregulated in prostate cancer cells, fueling tumor progression and disease aggressiveness. Different inhibitors, both synthetic and natural (highlighted in maroon boxes), have shown potential to target these metabolic vulnerabilities of dysregulated lipid metabolism and alleviate disease aggressiveness in prostate cancer. In addition, restoring LXR signaling through agonists/inducers (highlighted in dark green box) also offer promising approach to suppress dysregulated cholesterol metabolism and limit disease progression in prostate cancer.
Figure 3. Targeting dysregulated lipid metabolism in prostate cancer. Dsyregulated fatty acid and cholesterol metabolism often contribute to tumor progression and disease aggressiveness in prostate cancer. Various enzymes and transcription factors involved in this process, including FABPs, ACLY, ACAC, FASN, HMGCR, and SREBPs, are often dysregulated in prostate cancer cells, fueling tumor progression and disease aggressiveness. Different inhibitors, both synthetic and natural (highlighted in maroon boxes), have shown potential to target these metabolic vulnerabilities of dysregulated lipid metabolism and alleviate disease aggressiveness in prostate cancer. In addition, restoring LXR signaling through agonists/inducers (highlighted in dark green box) also offer promising approach to suppress dysregulated cholesterol metabolism and limit disease progression in prostate cancer.
None.
Ethical policy
Non applicable.
Availability of data and materials
All data generated or analysed during this study are included in this publication.
Author contributions
Ukasha Ahmed contributed to design of the work, data collection, and drafting the article.
Competing interests
The author declares no competing interests.
Funding
None.
- Bray F, Laversanne M, Sung H: Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2024, 74(3): 229-263.
- Siegel RL, Kratzer TB: Cancer statistics, 2025. CA Cancer J Clin 2025, 75(1):10-45.
- Parupathi P, Devarakonda LS: Reprogrammed Lipid Metabolism-Associated Therapeutic Vulnerabilities in Prostate Cancer. Int J Mol Sci 2025, 26(18): 9132.
- Halpern JA, Oromendia C, Shoag JE, Mittal S, Cosiano MF, Ballman KV, Vickers AJ, Hu JC: Use of Digital Rectal Examination as an Adjunct to Prostate Specific Antigen in the Detection of Clinically Significant Prostate Cancer. J Urol 2018, 199(4): 947-953.
- Achard V, Panje CM, Engeler D, Zilli T, Putora PM: Localized and Locally Advanced Prostate Cancer: Treatment Options. Oncology 2021, 99(7): 413-421.
- Moul JW: The changing face of castrate resistant prostate cancer. Prostate Cancer Prostatic Dis 2025, 28(3): 535-536.
- Zheng Z, Li J, Liu Y, Shi Z: The Crucial Role of AR-V7 in Enzalutamide-Resistance of Castration-Resistant Prostate Cancer. Cancers (Basel) 2022, 14(19): 4877.
- Dror CM, Chi KN: Apalutamide for the treatment of metastatic castration-sensitive prostate cancer. Future Oncol 2020, 16(35): 2905-2916.
- Armstrong AJ, Azad AA: Improved Survival With Enzalutamide in Patients With Metastatic Hormone-Sensitive Prostate Cancer. J Clin Oncol 2022, 40(15): 1616-1622.
- Faubert B, Solmonson A: Metabolic reprogramming and cancer progression. Science 2020, 368(6487): eaaw5473.
- Cardoso HJ, Carvalho TMA, Fonseca LRS, Figueira MI, Vaz CV, Socorro S: Revisiting prostate cancer metabolism: From metabolites to disease and therapy. Med Res Rev 2021, 41(3): 1499-1538.
- Pujana-Vaquerizo M, Bozal-Basterra L: Metabolic adaptations in prostate cancer. Br J Cancer 2024, 131(8): 1250-1262.
- Lima AR, Carvalho M: Comprehensive Metabolomics and Lipidomics Profiling of Prostate Cancer Tissue Reveals Metabolic Dysregulations Associated with Disease Development. J Proteome Res 2022, 21(3): 727-739.
- Butler LM, Perone Y, Dehairs J, Lupien LE, de Laat V, Talebi A, Loda M, Kinlaw WB, Swinnen JV: Lipids and cancer: Emerging roles in pathogenesis, diagnosis and therapeutic intervention. Adv Drug Deliv Rev 2020, 159: 245-293.
- Migita T, Takayama KI, Urano T, Obinata D, Ikeda K, Soga T, Takahashi S, Inoue S: ACSL3 promotes intratumoral steroidogenesis in prostate cancer cells. Cancer Sci 2017, 108(10): 2011-2021.
- Balaban S, Nassar ZD: Extracellular Fatty Acids Are the Major Contributor to Lipid Synthesis in Prostate Cancer. Mol Cancer Res 2019, 17(4): 949-962.
- Abdel-Wahab AF, Mahmoud W, Al-Harizy RM: Targeting glucose metabolism to suppress cancer progression: prospective of anti-glycolytic cancer therapy. Pharmacol Res 2019, 150: 104511.
- Liu Y, Sun Y, Guo Y, Shi X, Chen X, Feng W, Wu LL, Zhang J, Yu S, Wang Y et al: An Overview: The Diversified Role of Mitochondria in Cancer Metabolism. Int J Biol Sci 2023, 19(3): 897-915.
- Lu J, Tan M, Cai Q: The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett 2015, 356(2 Pt A): 156-164.
- Luo Y, Ma J, Lu W: The Significance of Mitochondrial Dysfunction in Cancer. Int J Mol Sci 2020, 21(16): 5598.
- Ahmad F, Cherukuri MK, Choyke PL: Metabolic reprogramming in prostate cancer. Br J Cancer 2021, 125(9): 1185-1196.
- Costello LC, Franklin RB: A comprehensive review of the role of zinc in normal prostate function and metabolism; and its implications in prostate cancer. Arch Biochem Biophys 2016, 611: 100-112.
- Cutruzzolà F, Giardina G, Marani M, Macone A, Paiardini A, Rinaldo S, Paone A: Glucose Metabolism in the Progression of Prostate Cancer. Front Physiol 2017, 8: 97.
- Acevedo S, Segovia MF, de la Fuente-Ortega E: Emerging Perspectives in Zinc Transporter Research in Prostate Cancer: An Updated Review. Nutrients 2024, 16(13): 2026.
- Lasorsa F, di Meo NA, Rutigliano M, Ferro M: Emerging Hallmarks of Metabolic Reprogramming in Prostate Cancer. Int J Mol Sci 2023, 24(2): 910.
- Liu J, Chen G, Liu Z, Liu S, Cai Z, You P, Ke Y, Lai L, Huang Y, Gao H et al: Aberrant FGFR Tyrosine Kinase Signaling Enhances the Warburg Effect by Reprogramming LDH Isoform Expression and Activity in Prostate Cancer. Cancer Res 2018, 78(16): 4459-4470.
- Xu L, Ma E, Zeng T, Zhao R, Tao Y, Chen X, Groth J, Liang C, Hu H, Huang J: ATM deficiency promotes progression of CRPC by enhancing Warburg effect. Endocr Relat Cancer 2019, 26(1): 59-71.
- Wang L, Wang J, Xiong H, Wu F, Lan T, Zhang Y, Guo X, Wang H, Saleem M, Jiang C et al: Co-targeting hexokinase 2-mediated Warburg effect and ULK1-dependent autophagy suppresses tumor growth of PTEN- and TP53-deficiency-driven castration-resistant prostate cancer. EBioMedicine 2016, 7: 50-61.
- Batchuluun B, Pinkosky SL, Steinberg GR: Lipogenesis inhibitors: therapeutic opportunities and challenges. Nat Rev Drug Discov 2022, 21(4): 283-305.
- Zhang Z, Wang W, Kong P, Feng K, Liu C, Sun T, Sang Y, Duan X, Tao Z, Liu W: New insights into lipid metabolism and prostate cancer (Review). Int J Oncol 2023, 62(6): 74.
- Wu X, Daniels G, Lee P, Monaco ME: Lipid metabolism in prostate cancer. Am J Clin Exp Urol 2014, 2(2): 111-120.
- Škara L, Huđek Turković A, Pezelj I, Vrtarić A, Sinčić N, Krušlin B, Ulamec M: Prostate Cancer-Focus on Cholesterol. Cancers (Basel) 2021, 13(18): 4696.
- Scaglia N, Frontini-López YR, Zadra G: Prostate Cancer Progression: as a Matter of Fats. Front Oncol 2021, 11: 719865.
- Watt MJ, Clark AK, Selth LA: Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci Transl Med 2019, 11(478): eaau5758.
- Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A, Hueto JA et al: Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541(7635): 41-45.
- Nath A, Chan C: Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Sci Rep 2016, 6: 18669.
- Kushwaha PP, Verma SS, Shankar E, Lin S, Gupta S: Role of solute carrier transporters SLC25A17 and SLC27A6 in acquired resistance to enzalutamide in castration-resistant prostate cancer. Mol Carcinog 2022, 61(4): 397-407.
- Olokpa E, Bolden A, Stewart LV: The Androgen Receptor Regulates PPARγ Expression and Activity in Human Prostate Cancer Cells. J Cell Physiol 2016, 231(12): 2664-2672.
- Fujita K, Kume H, Matsuzaki K, Kawashima A, Ujike T, Nagahara A, Uemura M, Miyagawa Y, Tomonaga T, Nonomura N: Proteomic analysis of urinary extracellular vesicles from high Gleason score prostate cancer. Sci Rep 2017, 7: 42961.
- Naeem AA, Abdulsamad SA, Zeng H, He G, Jin X, Zhang J, Alenezi BT, Ma H, Rudland PS, Ke Y: FABP5 can substitute for androgen receptor in malignant progression of prostate cancer cells. Int J Oncol 2024, 64(2): 18.
- Flaig TW, Salzmann-Sullivan M, Su LJ, Zhang Z, Joshi M, Gijón MA, Kim J, Arcaroli JJ, Van Bokhoven A, Lucia MS et al: Lipid catabolism inhibition sensitizes prostate cancer cells to antiandrogen blockade. Oncotarget 2017, 8(34): 56051-56065.
- Abudurexiti M, Zhu W, Wang Y, Wang J: Targeting CPT1B as a potential therapeutic strategy in castration-resistant and enzalutamide-resistant prostate cancer. Prostate 2020, 80(12): 950-961.
- Mah CY, Nguyen ADT, Niijima T, Helm M, Dehairs J: Peroxisomal β-oxidation enzyme, DECR2, regulates lipid metabolism and promotes treatment resistance in advanced prostate cancer. Br J Cancer 2024, 130(5): 741-754.
- Göbel A, Pählig S, Motz A, Breining D, Traikov S, Hofbauer LC, Rachner TD: Overcoming statin resistance in prostate cancer cells by targeting the 3-hydroxy-3-methylglutaryl-CoA-reductase. Biochem Biophys Res Commun 2024, 710: 149841.
- di Meo NA, Lasorsa F, Rutigliano M, Milella M, Ferro M, Battaglia M: The dark side of lipid metabolism in prostate and renal carcinoma: novel insights into molecular diagnostic and biomarker discovery. Expert Rev Mol Diagn 2023, 23(4): 297-313.
- Chimento A, Casaburi I, Avena P, Trotta F, De Luca A, Rago V, Pezzi V, Sirianni R: Cholesterol and Its Metabolites in Tumor Growth: Therapeutic Potential of Statins in Cancer Treatment. Front Endocrinol (Lausanne) 2018, 9: 807.
- Lee HJ, Li J, Vickman RE, Li J, Liu R, Durkes AC, Elzey BD, Yue S, Liu X, Ratliff TL et al: Cholesterol Esterification Inhibition Suppresses Prostate Cancer Metastasis by Impairing the Wnt/β-catenin Pathway. Mol Cancer Res 2018, 16(6): 974-985.
- Vaz CV, Marques R, Alves MG, Oliveira PF, Cavaco JE, Maia CJ, Socorro S: Androgens enhance the glycolytic metabolism and lactate export in prostate cancer cells by modulating the expression of GLUT1, GLUT3, PFK, LDH and MCT4 genes. J Cancer Res Clin Oncol 2016, 142(1): 5-16.
- Gonzalez-Menendez P, Hevia D, Rodriguez-Garcia A, Mayo JC, Sainz RM: Regulation of GLUT transporters by flavonoids in androgen-sensitive and -insensitive prostate cancer cells. Endocrinology 2014, 155(9): 3238-3250.
- Shafiee G, Saidijam M, Tayebinia H, Khodadadi I: Beneficial effects of genistein in suppression of proliferation, inhibition of metastasis, and induction of apoptosis in PC3 prostate cancer cells. Arch Physiol Biochem 2022, 128(3): 694-702.
- Song YY, Yuan Y, Shi X, Che YY: Improved drug delivery and anti-tumor efficacy of combinatorial liposomal formulation of genistein and plumbagin by targeting Glut1 and Akt3 proteins in mice bearing prostate tumor. Colloids Surf B Biointerfaces 2020, 190: 110966.
- Tang Q, Ma J, Sun J, Yang L, Yang F, Zhang W, Li R, Wang L, Wang Y, Wang H: Genistein and AG1024 synergistically increase the radiosensitivity of prostate cancer cells. Oncol Rep 2018, 40(2): 579-588.
- Wang J, Xu W, Wang B, Lin G, Wei Y, Abudurexiti M, Zhu W, Liu C, Qin X, Dai B et al: GLUT1 is an AR target contributing to tumor growth and glycolysis in castration-resistant and enzalutamide-resistant prostate cancers. Cancer Lett 2020, 485: 45-55.
- Saxena R, Yang C, Rao M, Turaga RC, Garlapati C, Gundala SR, Myers K, Ghareeb A, Bhattarai S, Kamalinia G et al: Preclinical Development of a Nontoxic Oral Formulation of Monoethanolamine, a Lipid Precursor, for Prostate Cancer Treatment. Clin Cancer Res 2017, 23(14): 3781-3793.
- Garlapati C, Joshi S, Turaga RC, Mishra M, Reid MD, Kapoor S, Artinian L, Rehder V, Aneja R: Monoethanolamine-induced glucose deprivation promotes apoptosis through metabolic rewiring in prostate cancer. Theranostics 2021, 11(18): 9089-9106.
- Park Y, Lee HJ, Sim DY, Park JE, Ahn CH, Park SY, Lee YC, Shim BS, Kim B, Kim SH: Inhibition of glycolysis and SIRT1/GLUT1 signaling ameliorates the apoptotic effect of Leptosidin in prostate cancer cells. Phytother Res 2024, 38(3): 1235-1244.
- Ciscato F, Ferrone L: Hexokinase 2 in Cancer: A Prima Donna Playing Multiple Characters. Int J Mol Sci 2021, 22(9): 4716 .
- Stein M, Lin H, Jeyamohan C, Dvorzhinski D, Gounder M, Bray K, Eddy S, Goodin S, White E, Dipaola RS: Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate 2010, 70(13): 1388-1394.
- Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, Le Marchand-Brustel Y, Giorgetti-Peraldi S, Cormont M, Bertolotto C et al: Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res 2010, 70(6): 2465-2475.
- Zhang H, Du X, Sun TT, Wang CL, Li Y, Wu SZ: Lectin PCL inhibits the Warburg effect of PC3 cells by combining with EGFR and inhibiting HK2. Oncol Rep 2017, 37(3): 1765-1771.
- Hong T, Ham J, Song J, Song G: Brassinin Inhibits Proliferation in Human Liver Cancer Cells via Mitochondrial Dysfunction. Cells 2021, 10(2): 332.
- Jones BC, Pohlmann PR, Clarke R, Sengupta S: Treatment against glucose-dependent cancers through metabolic PFKFB3 targeting of glycolytic flux. Cancer Metastasis Rev 2022, 41(2): 447-458.
- Ren JG, Seth P, Ye H, Guo K, Hanai JI, Husain Z, Sukhatme VP: Citrate Suppresses Tumor Growth in Multiple Models through Inhibition of Glycolysis, the Tricarboxylic Acid Cycle and the IGF-1R Pathway. Sci Rep 2017, 7(1): 4537.
- Klarer AC, O'Neal J, Imbert-Fernandez Y, Clem A, Ellis SR, Clark J, Clem B, Chesney J, Telang S: Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism. Cancer Metab 2014, 2(1): 2.
- Dong XM, Chen L, Wu P, Cheng LH, Wang Y, Yang Y, Zhang Y, Tang WY, Xie T, Zhou JL: Targeted metabolomics reveals PFKFB3 as a key target for elemene-mediated inhibition of glycolysis in prostate cancer cells. Phytomedicine 2024, 123: 155185.
- Kim GJ, Jo HJ, Lee KJ, Choi JW, An JH: Oleanolic acid induces p53-dependent apoptosis via the ERK/JNK/AKT pathway in cancer cell lines in prostatic cancer xenografts in mice. Oncotarget 2018, 9(41): 26370-26386.
- Markowitsch SD, Juetter KM, Schupp P, Hauschulte K, Vakhrusheva O, Slade KS, Thomas A, Tsaur I: Shikonin Reduces Growth of Docetaxel-Resistant Prostate Cancer Cells Mainly through Necroptosis. Cancers (Basel) 2021, 13(4): 882.
- Wang L, Stadlbauer B, Lyu C, Buchner A, Pohla H: Shikonin enhances the antitumor effect of cabazitaxel in prostate cancer stem cells and reverses cabazitaxel resistance by inhibiting ABCG2 and ALDH3A1. Am J Cancer Res 2020, 10(11): 3784-3800.
- Tseng JC, Lin CY, Su LC, Fu HH, Yang SD, Chuu CP: CAPE suppresses migration and invasion of prostate cancer cells via activation of non-canonical Wnt signaling. Oncotarget 2016, 7(25): 38010-38024.
- Pak JN, Lee HJ, Sim DY, Park JE, Ahn CH, Park SY, Khil JH, Shim B, Kim B, Kim SH: Anti-Warburg effect via generation of ROS and inhibition of PKM2/β-catenin mediates apoptosis of lambertianic acid in prostate cancer cells. Phytother Res 2023, 37(9): 4224-4235.
- Sharma D, Singh M, Rani R: Role of LDH in tumor glycolysis: Regulation of LDHA by small molecules for cancer therapeutics. Semin Cancer Biol 2022, 87: 184-195.
- Xian ZY, Liu JM, Chen QK, Chen HZ, Ye CJ, Xue J, Yang HQ, Li JL, Liu XF, Kuang SJ: Inhibition of LDHA suppresses tumor progression in prostate cancer. Tumour Biol 2015, 36(10): 8093-8100.
- Lee YJ, Lee SH: ERK1/2-Dependent Inhibition of Glycolysis in Curcumin-Induced Cytotoxicity of Prostate Carcinoma Cells. biomed Res Int 2022, 2022: 7626405.
- Panahizadeh R, Vatankhah MA, Jeddi F, Arabzadeh A, Nejati-Koshki K, Salimnejad R, Najafzadeh N: Cytotoxicity of curcumin against CD44(±) prostate cancer cells: Roles of miR-383 and miR-708. Avicenna J Phytomed 2023, 13(4): 429-441.
- Cakici C, Daylan B, Unluer RS, Emekli-Alturfan E, Ayla S, Gozel HE, Yigit P, Dokgoz EY, Yigitbasi T: LDH-A Inhibitor as a Remedy to Potentiate the Anticancer Effect of Docetaxel in Prostate Cancer. J Cancer 2024, 15(3): 590-602.
- Woolbright BL, Rajendran G, Harris RA, Taylor JA, 3rd: Metabolic Flexibility in Cancer: Targeting the Pyruvate Dehydrogenase Kinase:Pyruvate Dehydrogenase Axis. Mol Cancer Ther 2019, 18(10): 1673-1681.
- Nunes-Xavier CE, Mingo J, Emaldi M, Flem-Karlsen K, Mælandsmo GM, Fodstad Ø, Llarena R, López JI, Pulido R: Heterogeneous Expression and Subcellular Localization of Pyruvate Dehydrogenase Complex in Prostate Cancer. Front Oncol 2022, 12: 873516.
- Zhang W, Zhang SL, Hu X, Tam KY: Phenyl butyrate inhibits pyruvate dehydrogenase kinase 1 and contributes to its anti-cancer effect. Eur J Pharm Sci 2017, 110: 93-100.
- Guan S, Zhao L, Peng R: Mitochondrial Respiratory Chain Supercomplexes: From Structure to Function. Int J Mol Sci 2022, 23(22): 13880.
- Okoye CN, Koren SA, Wojtovich AP: Mitochondrial complex I ROS production and redox signaling in hypoxia. Redox Biol 2023, 67: 102926.
- Chang E, Liu H, Unterschemmann K, Ellinghaus P, Liu S, Gekeler V, Cheng Z, Berndorff D, Gambhir SS: 18F-FAZA PET imaging response tracks the reoxygenation of tumors in mice upon treatment with the mitochondrial complex I inhibitor BAY 87-2243. Clin Cancer Res 2015, 21(2): 335-346.
- Qu Z, Hao F: Targeted Metabolic Alterations in Prostate Cancer: A Promising Therapeutic Strategy. Mol Pharm 2025, 22(9): 5212-5226.
- Dong LF, Low P, Dyason JC, Wang XF, Prochazka L, Witting PK, Freeman R, Swettenham E, Valis K, Liu J et al: Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene 2008, 27(31): 4324-4335.
- Fiorillo M, Lamb R, Tanowitz HB, Mutti L, Krstic-Demonacos M, Cappello AR, Martinez-Outschoorn UE, Sotgia F, Lisanti MP: Repurposing atovaquone: targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget 2016, 7(23): 34084-34099.
- Cooper CE, Brown GC: The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr 2008, 40(5): 533-539.
- Li B, Yu Y, Jiang Y, Zhao L, Li A, Li M, Yuan B, Lu J, Dong Z, Zhao J et al: Cloperastine inhibits esophageal squamous cell carcinoma proliferation in vivo and in vitro by suppressing mitochondrial oxidative phosphorylation. Cell Death Discov 2021, 7(1): 166.
- Ruas JS, Siqueira-Santos ES, Rodrigues-Silva E, Castilho RF: High glycolytic activity of tumor cells leads to underestimation of electron transport system capacity when mitochondrial ATP synthase is inhibited. Sci Rep 2018, 8(1): 17383.
- Al-Jameel W, Gou X, Forootan SS, Al Fayi MS, Rudland PS, Forootan FS, Zhang J, Cornford PA, Hussain SA, Ke Y: Inhibitor SBFI26 suppresses the malignant progression of castration-resistant PC3-M cells by competitively binding to oncogenic FABP5. Oncotarget 2017, 8(19): 31041-31056.
- Carbonetti G, Converso C, Clement T, Wang C, Trotman LC, Ojima I, Kaczocha M: Docetaxel/cabazitaxel and fatty acid binding protein 5 inhibitors produce synergistic inhibition of prostate cancer growth. Prostate 2020, 80(1): 88-98.
- Abdulsamad SA, Naeem AA, Zeng H, He G, Jin X, Alenezi BA, Ai J, Zhang J, Ma H, Rudland PS et al: Experimental treatment efficacy of dmrFABP5 on prostate cancer singly or in combination with drugs in use. Am J Cancer Res 2024, 14(1): 300-323.
- Khwairakpam AD, Shyamananda MS, Sailo BL, Rathnakaram SR, Padmavathi G, Kotoky J, Kunnumakkara AB: ATP citrate lyase (ACLY): a promising target for cancer prevention and treatment. Curr Drug Targets 2015, 16(2): 156-163.
- Shah S, Carriveau WJ, Li J, Campbell SL, Kopinski PK, Lim HW, Daurio N, Trefely S, Won KJ, Wallace DC et al: Targeting ACLY sensitizes castration-resistant prostate cancer cells to AR antagonism by impinging on an ACLY-AMPK-AR feedback mechanism. Oncotarget 2016, 7(28): 43713-43730.
- Gao Y, Islam MS, Tian J, Lui VW, Xiao D: Inactivation of ATP citrate lyase by Cucurbitacin B: A bioactive compound from cucumber, inhibits prostate cancer growth. Cancer Lett 2014, 349(1): 15-25.
- Liu S, Lai J, Feng Y, Zhuo Y, Zhang H, Chen Y, Li J, Mei X, Zeng Y, Su J et al: Acetyl-CoA carboxylase 1 depletion suppresses de novo fatty acid synthesis and mitochondrial β-oxidation in castration-resistant prostate cancer cells. J Biol Chem 2023, 299(1): 102720.
- Sadowski MC, Pouwer RH, Gunter JH, Lubik AA, Quinn RJ, Nelson CC: The fatty acid synthase inhibitor triclosan: repurposing an anti-microbial agent for targeting prostate cancer. Oncotarget 2014, 5(19): 9362-9381.
- Singh KB, Kim SH, Hahm ER, Pore SK, Jacobs BL, Singh SV: Prostate cancer chemoprevention by sulforaphane in a preclinical mouse model is associated with inhibition of fatty acid metabolism. Carcinogenesis 2018, 39(6): 826-837.
- Zadra G, Ribeiro CF, Chetta P, Ho Y, Cacciatore S, Gao X, Syamala S, Bango C, Photopoulos C, Huang Y et al: Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc Natl Acad Sci U S A 2019, 116(2): 631-640.
- Heuer TS, Ventura R, Mordec K, Lai J, Fridlib M, Buckley D, Kemble G: FASN Inhibition and Taxane Treatment Combine to Enhance Anti-tumor Efficacy in Diverse Xenograft Tumor Models through Disruption of Tubulin Palmitoylation and Microtubule Organization and FASN Inhibition-Mediated Effects on Oncogenic Signaling and Gene Expression. EBioMedicine 2017, 16: 51-62.
- Nambiar DK, Deep G, Singh RP, Agarwal C, Agarwal R: Silibinin inhibits aberrant lipid metabolism, proliferation and emergence of androgen-independence in prostate cancer cells via primarily targeting the sterol response element binding protein 1. Oncotarget 2014, 5(20): 10017-10033.
- Shao W, Machamer CE, Espenshade PJ: Fatostatin blocks ER exit of SCAP but inhibits cell growth in a SCAP-independent manner. J Lipid Res 2016, 57(8): 1564-1573.
- Chen M, Zhang J, Sampieri K, Clohessy JG, Mendez L, Gonzalez-Billalabeitia E, Liu XS, Lee YR, Fung J, Katon JM et al: An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat Genet 2018, 50(2): 206-218.
- Li X, Wu JB, Chung LW, Huang WC: Anti-cancer efficacy of SREBP inhibitor, alone or in combination with docetaxel, in prostate cancer harboring p53 mutations. Oncotarget 2015, 6(38): 41018-41032.
- Kim SH, Singh KB, Hahm ER, Lokeshwar BL, Singh SV: Withania somnifera root extract inhibits fatty acid synthesis in prostate cancer cells. J Tradit Complement Med 2020, 10(3): 188-197.
- Hsieh PF, Jiang WP, Basavaraj P, Huang SY, Ruangsai P, Wu JB, Huang GJ, Huang WC: Cell suspension culture extract of Eriobotrya japonica attenuates growth and induces apoptosis in prostate cancer cells via targeting SREBP-1/FASN-driven metabolism and AR. Phytomedicine 2021, 93: 153806.
- Huang SY, Huang GJ, Wu HC, Kao MC, Huang WC: Ganoderma tsugae Inhibits the SREBP-1/AR Axis Leading to Suppression of Cell Growth and Activation of Apoptosis in Prostate Cancer Cells. Molecules 2018, 23(10): 2539.
- Wei G, Huang Y, Li W, Xie Y, Zhang D, Niu Y: SREBF1-based metabolic reprogramming in prostate cancer promotes tumor ferroptosis resistance. Cell Death Dis 2025, 11(1): 75.
- Guerrero-Ochoa P, Rodríguez-Zapater S: Prostate Cancer and the Mevalonate Pathway. Int J Mol Sci 2024, 25(4): 2152.
- Kochuparambil ST, Al-Husein B, Goc A, Soliman S, Somanath PR: Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression. J Pharmacol Exp Ther 2011, 336(2): 496-505.
- Bilotta MT, Petillo S, Santoni A, Cippitelli M: Liver X Receptors: Regulators of Cholesterol Metabolism, Inflammation, Autoimmunity, and Cancer. Front Immunol 2020, 11: 584303.
- Chen T, Xu J, Fu W: EGFR/FOXO3A/LXR-α Axis Promotes Prostate Cancer Proliferation and Metastasis and Dual-Targeting LXR-α/EGFR Shows Synthetic Lethality. Front Oncol 2020, 10: 1688.
Annals of urologic oncology
p-ISSN: 2617-7765, e-ISSN: 2617-7773
 Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.