
 Submit Manuscript
Submit Manuscript
Case Report | Open Access
Incidentally Discovered Primary Renal Leiomyosarcoma in a Middle Aged Female Presenting with Symptoms of Incomplete Abortion: A Rare Case Report
Rashi Maheshwari1, Shikhar Chohan1, Ayushi Agarwal1, Sufian Zaheer11Department of Pathology, Vardhman Mahavir Medical College and Safdarjung Hospital, New Delhi, India.
Correspondence: Sufian Zaheer (Department of Pathology, Vardhman Mahavir Medical College and Safdarjung Hospital, New Delhi, India; Email: sufianzaheer@gmail.com).
Annals of Urologic Oncology 2025, 8(3): 116-121. https://doi.org/10.32948/auo.2025.09.04
Received: 28 Aug 2025 | Accepted: 11 Sep 2025 | Published online: 23 Sep 2025
Case Presentation We describe a 37-year-old woman, notably younger than the typical age reported for renal LMS, who presented with abnormal uterine bleeding and incomplete abortion. During gynaecologic imaging, an incidental exophytic right-renal mass was detected. CECT and CT angiography revealed a heterogeneously enhancing lesion with necrosis abutting adjacent structures. The patient underwent laparoscopic radical nephrectomy. Histopathological examination showed fascicles of spindle cells with pleomorphism, atypical mitoses, and necrosis involving <50% of tumor area. Immunohistochemistry confirmed smooth muscle differentiation with positivity for vimentin, desmin, and SMA, while negative for cytokeratin, S100, and HMB-45. A final diagnosis of well-differentiated primary renal leiomyosarcoma (FNCLCC grade 1, stage pT3aNxMx) was rendered. The postoperative course was uneventful, and the patient remains well at 3-month follow-up.
Conclusion This case is novel for its incidental detection during gynaecologic evaluation for incomplete abortion and for occurring in a young female patient (37 years), whereas most reported renal LMS present later in life and at higher grade. These features underscore the importance of considering LMS in atypical contexts and highlight the need for awareness among both urologists and gynaecologists.
Key words renal leiomyosarcoma, primary renal sarcoma, kidney neoplasm, smooth muscle tumor, radical nephrectomy, case report
To add to the current understating of renal LMS, we present a case of incidentally discovered primary renal LMS in a middle-aged female.
A 37-year-old lady presented to gynaecology OPD with chief complaints of abnormal bleeding per vaginum for a period of 5 days. On local examination, external genitalia was unremarkable and external os was visible and unremarkable. On per abdomen examination, no significant abnormality was detected. Preliminary blood investigations were within normal limits.
Imaging
On further workup, a whole abdomen ultrasonography was performed. On USG, the uterus was retroverted, normal in size with presence of heterogenous echotexture with few well defined heterogenous hypoechoic lesions, largest in anterior myometrium measuring 8x6 mm, suggestive of multiple leiomyomas. Also seen was ill defined heterogenous hyperechoic lesion measuring 9.1x7.9 mm, within the endometrial cavity showing internal vascularity, suggestive of retained products of conception (likely the cause of abnormal uterine bleeding in the patient) for which a hysteroscopic D&C was done with Histopathological diagnosis of retained products of gestation. Further in the same USG, urinary bladder and left kidney were unremarkable on USG. However, right kidney was slightly enlarged in size and showed an exophytic heterogenous echoic lesion arising from the mid pole, extending and abutting the gall bladder and liver with mild internal vascularity and measuring 74x51 mm.
Further radiological investigations were done to find the nature and extent of the lesion in the kidney. Contrast enhanced CT scan showed the exophytic mass to be arising from the anterior mid pole of the right kidney, significantly extending anteriorly with loss of fat planes with gall bladder, second part of duodenum and adjacent segment V of liver. The findings were suggestive of neoplastic etiology of right kidney (Figure 1).
CT renal angiography was performed which revealed similar radiological findings. Further, areas of necrosis were noted. The mass was seen bulging into the perinephric fat and extending superiorly. However, no extension was seen beyond gerota’s fascia. The mass was indenting the right renal vessels with contact angle of 180 degrees. Right renal vein and IVC were normal. Ipsilateral adrenal gland was also unremarkable (Figure 2).
Surgical management
The patient was planned for laparoscopic radical nephrectomy of right kidney under general anesthesia. Intra operative findings revealed right kidney to be grossly enlarged involving the upper and mid pole and adherent to 2nd part of duodenum. One renal artery and 2 renal veins were clipped and divided. No liver, visceral and peritoneal metastasis were seen. The specimen was sent for histopathological examination.
Histopathological examination
On gross examination, the right kidney measured 11x10x5 cm and weighed approximately 500gm. A mass was identified involving the upper and middle pole of the kidney and measuring 7x6x5cm. Capsular breach was identified, and the mass was seen extending to perinephric fat. The mass macroscopically seemed to arise from renal pelvis. Hilar structures could not be identified. On serial sectioning, corticomedullary junction was identified. Tumor on serial sectioning appeared solid white in appearance with hemorrhagic areas. On microscopic examination, the tumor was composed of plump spindle cells arranged in bundles and long intersecting fascicles, with individual cells having oval to elongated, blunt-ended nuclei and moderate to abundant pale to eosinophilic cytoplasm, showing moderate to marked nuclear pleomorphism. Mitotic figures including atypical ones were numerous (Figure 3). Tumor necrosis was seen involving <50% of the total tumor area. Lymphatic or vascular invasion were not identified. Margins of resection were free of tumor. On immunohistochemistry, the tumor cells were positive for vimentin, desmin and smooth muscle actin and negative for CK7, PanCK, MyoD1, S100, p53, CD10, HMB-45 and EMA (Figure 4). Histopathological reporting was done according to CAP cancer templates [7]. Based on the morphological and immunohistochemical profile, a diagnosis of Well-differentiated leiomyosarcoma was made. The tumor was involving the upper and middle pole of right total nephrectomy specimen and was involving the perinephric fat and reaching upto the gerota’s fascia. The pelvic calyceal system was seen involved by the tumor, however renal sinus was free of tumor. Final diagnosis was given as Leiomyosarcoma, FNCLCC grade 1, of right kidney with pTNM staging as pT3aNxMx. No regional lymph nodes were submitted.
Follow up
Post op period was uneventful. Urinary catheter was removed on post op day 1 and drain was removed on post op day 2. At present, the patient has been followed up for 3 months, during which she has remained asymptomatic with no clinical or radiological evidence of recurrence or metastasis. The patient has been counselled regarding the importance of regular clinical review and imaging, and she has been enrolled in our institutional sarcoma follow-up program. The planned protocol includes physical examination and abdominal imaging (ultrasound or CT) every 6 months for the first 2–3 years, in addition to annual chest imaging to monitor for pulmonary metastasis. After this period, surveillance will be continued on an annual basis or sooner if new symptoms develop.
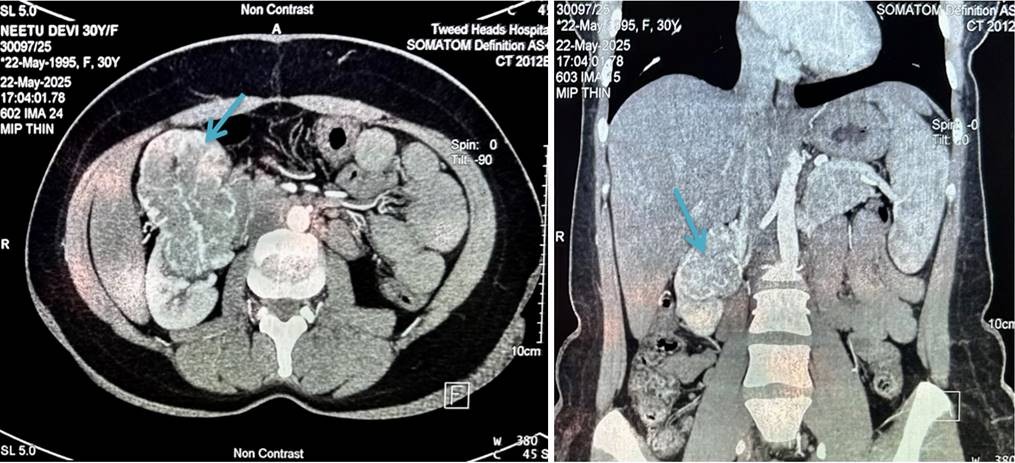 Figure 1. CECT Abdomen – An approximate 75 x 54 mm size exophytic enhancing soft tissue mass lesion is seen arising from the anterior mid pole of right kidney, significantly extending anteriorly and having lost fat plane with gall bladder, 2nd part of duodenum and adjacent segment V of liver. Findings were suggestive of neoplastic aetiology of right kidney. Another 16 x 12mm size soft tissue lesion is seen at mid pole of right kidney just above the large lesion likely neoplastic aetiology.
Figure 1. CECT Abdomen – An approximate 75 x 54 mm size exophytic enhancing soft tissue mass lesion is seen arising from the anterior mid pole of right kidney, significantly extending anteriorly and having lost fat plane with gall bladder, 2nd part of duodenum and adjacent segment V of liver. Findings were suggestive of neoplastic aetiology of right kidney. Another 16 x 12mm size soft tissue lesion is seen at mid pole of right kidney just above the large lesion likely neoplastic aetiology.
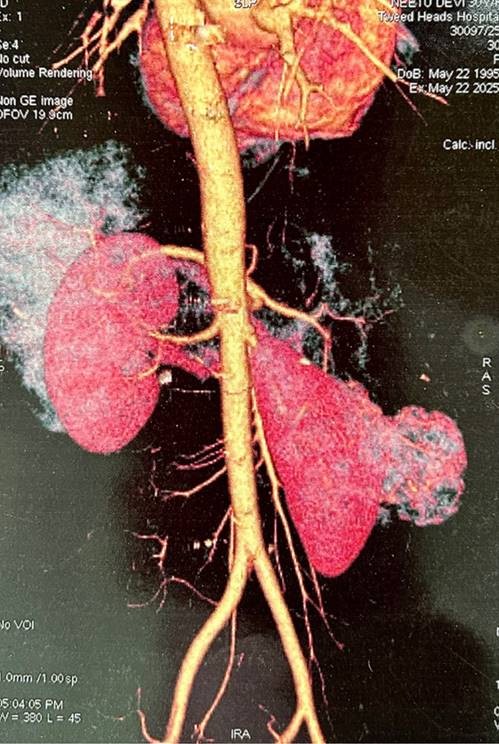 Figure 2. CT renal angiography – Large well defined smoothly marginated heterogenously enhancing exophytic mass is seen arising from mid pole of right kidney measuring “7.4 x 5.1 cm” with necrotic areas(white arrow). No calcification is seen. No fat density is seen. It is bulging into perinephric fat and extending superiorly. No extension seen beyond Gerota’s fascia. Right renal vein and IVC are normal. Ipsilateral left adrenal gland is normal. It is abutting inferior surface of right lobe. It is indenting right renal vessels with contact angle of 180°.
Figure 2. CT renal angiography – Large well defined smoothly marginated heterogenously enhancing exophytic mass is seen arising from mid pole of right kidney measuring “7.4 x 5.1 cm” with necrotic areas(white arrow). No calcification is seen. No fat density is seen. It is bulging into perinephric fat and extending superiorly. No extension seen beyond Gerota’s fascia. Right renal vein and IVC are normal. Ipsilateral left adrenal gland is normal. It is abutting inferior surface of right lobe. It is indenting right renal vessels with contact angle of 180°.
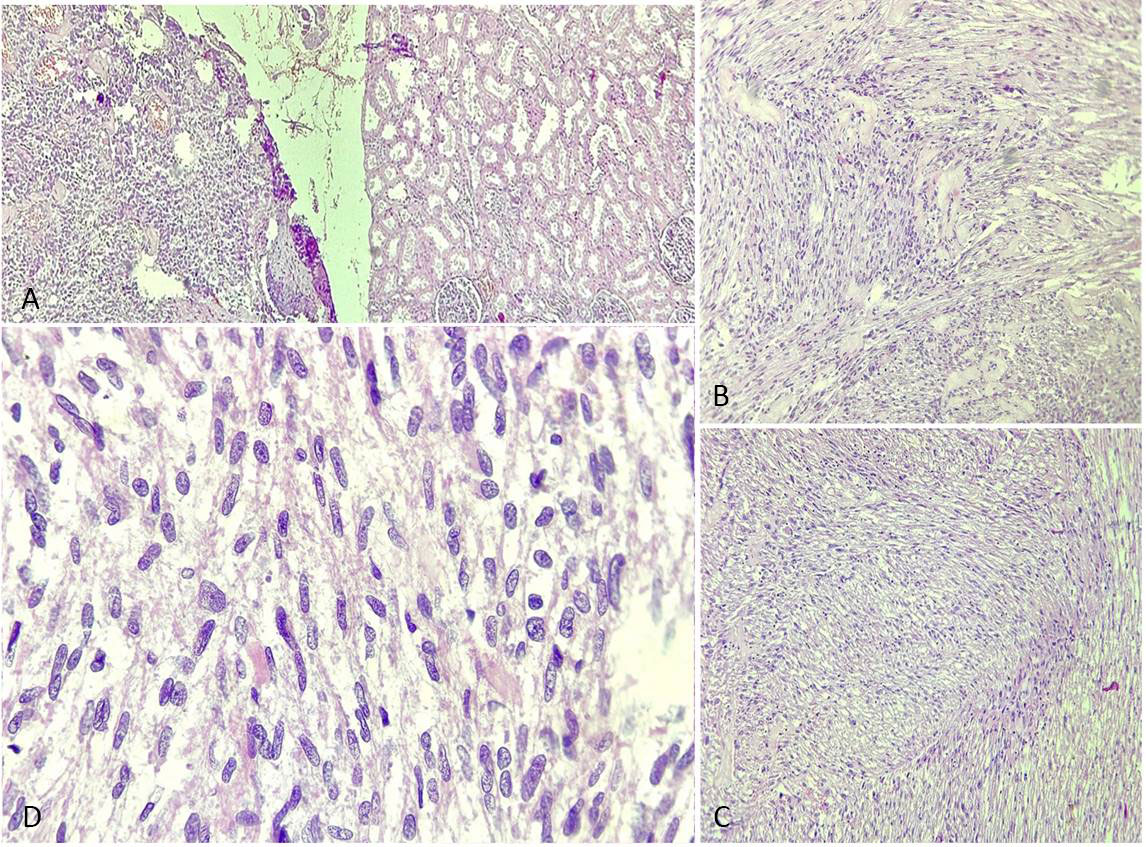 Figure 3. Renal leiomyosarcoma (H&E stained sections). (a) Tumor cells (black arrow) in fascicles adjacent to normal renal tissue ( blue arrow), H&E, 4×; (b-c) Fascicles of spindle cells (black arrow) with oval to elongated, blunt-ended nuclei and moderate eosinophilic cytoplasm, H&E, 10×; (d) High power showing marked nuclear pleomorphism and atypical mitoses (black arrow), H&E, 40×.
Figure 3. Renal leiomyosarcoma (H&E stained sections). (a) Tumor cells (black arrow) in fascicles adjacent to normal renal tissue ( blue arrow), H&E, 4×; (b-c) Fascicles of spindle cells (black arrow) with oval to elongated, blunt-ended nuclei and moderate eosinophilic cytoplasm, H&E, 10×; (d) High power showing marked nuclear pleomorphism and atypical mitoses (black arrow), H&E, 40×.
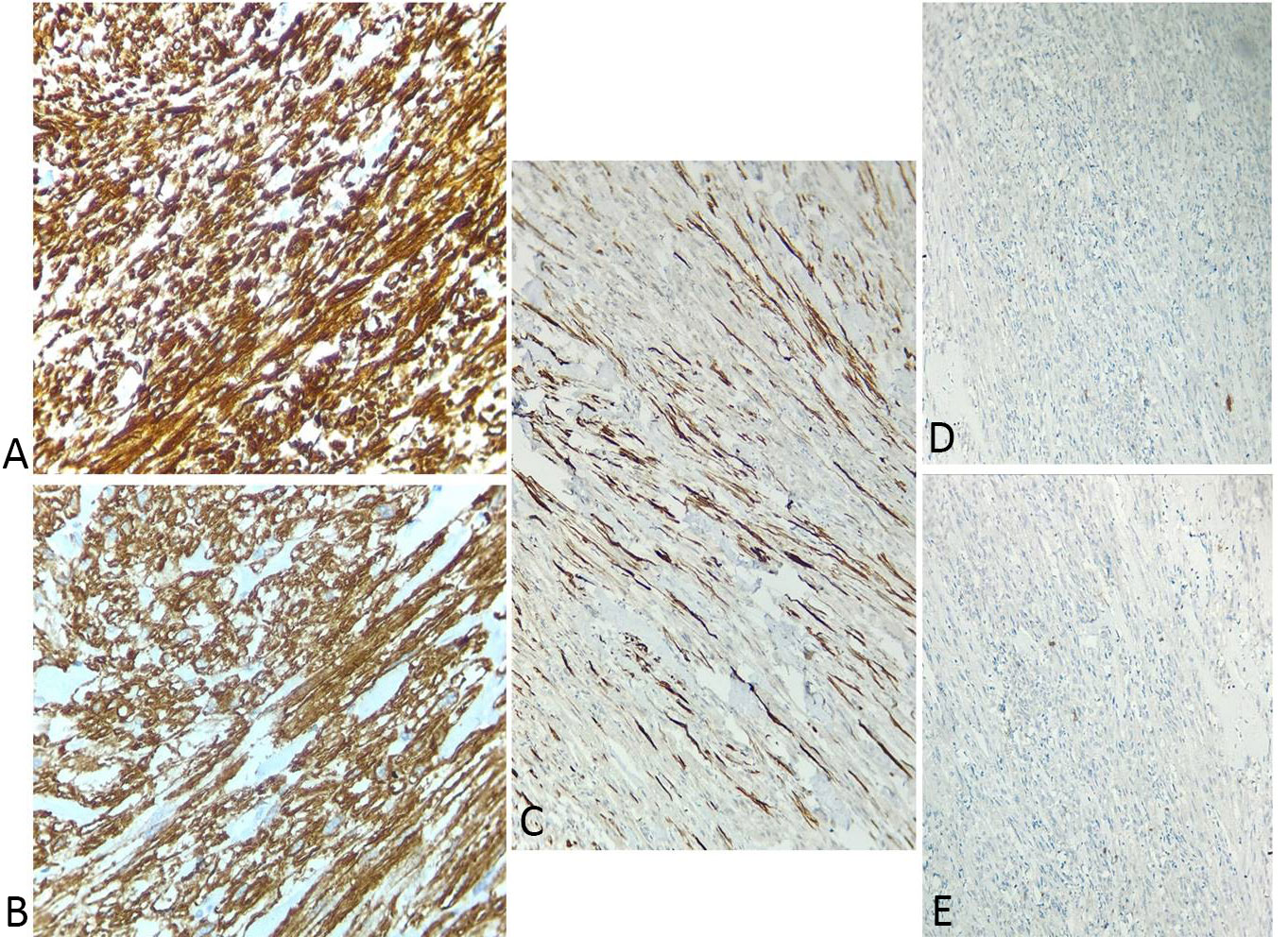 Figure 4. Immunohistochemistry of renal leiomyosarcoma. (a) Strong cytoplasmic positivity for Vimentin (clone V9, Dako), 40×; (b) Diffuse cytoplasmic positivity for SMA (clone 1A4, Dako), 40×; (c) Cytoplasmic positivity for Desmin (clone D33, Dako), 40×; (d) Tumor cells negative for MyoD1 (clone 5.8A, Dako), 10×; (e) Tumor cells negative for Pan-CK (clone AE1/AE3, Dako), 10×.
Figure 4. Immunohistochemistry of renal leiomyosarcoma. (a) Strong cytoplasmic positivity for Vimentin (clone V9, Dako), 40×; (b) Diffuse cytoplasmic positivity for SMA (clone 1A4, Dako), 40×; (c) Cytoplasmic positivity for Desmin (clone D33, Dako), 40×; (d) Tumor cells negative for MyoD1 (clone 5.8A, Dako), 10×; (e) Tumor cells negative for Pan-CK (clone AE1/AE3, Dako), 10×.
No applicable.
Ethical policy
The investigation was conducted in accordance with the Declaration of Helsinki of 1975. Written informed consent was taken from the patient.
Availability of data and materials
Data sharing is not applicable to this article as no new data were created or analysed in this study.
Author contributions
Rashi Maheshwari: Data Curation, Visualization, Writing - Original Draft; Shikhar Chohan: Data Curation, Visualization, Writing - Original Draft; Ayushi Aggarwal: Data Curation, Visualization; Sufian Zaheer: Conceptualization, Data Curation, Validation, Formal analysis, Writing - Review & Editing, Visualization.
Competing interests
The authors declare that they have no competing interests.
Funding
NIL.
- BlueBooksOnline. Who.int. Available from: https://tumourclassification.iarc.who.int/chapters/36
- Padala SA, Barsouk A, Thandra KC, Saginala K, Mohammed A, Vakiti A, Rawla P, Barsouk A: Epidemiology of renal cell carcinoma. World J Oncol 2020, 11(3): 79-87.
- Chacko A, Gopinathan V, Roy S, Prashanty A, Kumar S: 458 primary renal mesenchymal neoplasms: A retrospective clinicopathological study. Br J Surg 2023, 110(Supplement_7).
- Dotan ZA, Tal R, Golijanin D, Snyder ME, Antonescu C, Brennan MF, Russo P: Adult genitourinary sarcoma: the 25-year memorial sloan-kettering experience. J Urol 2006, 176(5): 2033-2039.
- Mondaini N, Palli D, Saieva C, Nesi G, Franchi A, Ponchietti R, Tripodi S, Miracco C, Meliani E, Carini M et al: Clinical characteristics and overall survival in genitourinary sarcomas treated with curative intent: a multicenter study. Eur Urol 2005, 47(4): 468-473.
- Beardo P, José Ledo M, Ruiz Campos JL: Renal leiomyosarcoma. Rare Tumors 2013, 5(3): 144-145.
- Cancer Protocol Templates [Internet]. College of American Pathologists. 2017.
- Trojani M, Contesso G, Coindre JM, Rouesse J, Bui NB, de Mascarel A, Goussot JF, David M, Bonichon F, Lagarde C: Soft-tissue sarcomas of adults; study of pathological prognostic variables and definition of a histopathological grading system. Int J Cancer 1984, 33(1): 37-42.
- The International Agency for Research on Cancer (IARC). Global Cancer Observatory.
- Gupta D, Singh A, Gupta N, Mehra N, Bahuguna P, Aggarwal V, Krishnamurthy MN, Roy PS, Malhotra P, Gupta S et al: Cost-effectiveness of the first line treatment options for metastatic renal cell carcinoma in India. JCO Glob Oncol 2023, 9: e2200246.
- Periasamy K, Dey T, Goyal S, Madan R, Kumar S, Devana SK, Elumalai T, Giridhar P, Ghoshal S, Kapoor R et al: Primary renal leiomyosarcoma in adult patients: a systematic review and individual patient data analysis. Afr J Urol 2024, 30(1): 15.
- Miller JS, Zhou M, Brimo F, Guo CC, Epstein JI: Primary leiomyosarcoma of the kidney: a clinicopathologic study of 27 cases. Am J Surg Pathol 2010, 34(2): 238-242.
- Novak M, Perhavec A, Maturen KE, Pavlovic Djokic S, Jereb S, Erzen D: Leiomyosarcoma of the renal vein: analysis of outcome and prognostic factors in the world case series of 67 patients. Radiol Oncol 2017, 51(1): 56-64.
- Dhawan S, Chopra P, Dhawan S: Primary renal leiomyosarcoma: A diagnostic challenge. Urol Ann 2012, 4(1): 48-50.
- Öztürk H: Prognostic features of renal sarcomas (Review). Oncol Lett 2015, 9(3): 1034-1038.
- Sood N, Varshney N, Rawat A: Cytohistomorphological features of primary renal leiomyosarcoma: A rare case report. Int J Res Rev 2025, 12(6): 301-305.
- Ding Y, Quan C: Primary renal leiomyosarcoma. J Coll Physicians Surg Pak 2021, 31(6): 725-727.
- Zafar R, Manthri S, Shurbaji MS: Renal leiomyosarcoma. In: StatPearls. Treasure Island (FL): StatPearls Publishing, 2025.
- Karaosmanoğlu AD, Onur MR, Shirkhoda A, Ozmen M, Hahn PF: Unusual Malignant Solid Neoplasms of the Kidney: Cross-Sectional Imaging Findings. Korean J Radiol 2015, 16(4): 853-859.
- Deyrup AT, Montgomery E, Fisher C: Leiomyosarcoma of the kidney: a clinicopathologic study. Am J Surg Pathol 2004, 28(2): 178-182.
- Demir A, Yazici CM, Eren F, Türkeri L: Case report: good prognosis in leiomyosarcoma of the kidney. Int Urol Nephrol 2007, 39(1): 7-10.
- Srinivasan S, Paul A, Pravallika H, Mekala IN, Sriramaneni VR: Primary Renal Leiomyosarcoma: A Case Report of a Rare and Aggressive Neoplasm. Cureus 2025, 17(7): e87099.
- Moazzam M, Ather MH, Hussainy AS: Leiomyosarcoma presenting as a spontaneously ruptured renal tumor-case report. BMC Urol 2002, 2(1): 13.
Annals of urologic oncology
p-ISSN: 2617-7765, e-ISSN: 2617-7773
 Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.