
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
Metabolic Reprogramming in Kidney Cancer: Implications for Therapy
Elena Tena Edo11Faculty of Health Sciences, International University of La Rioja, Av. de la Paz, 137, 26006 Logrono, La Rioja, Spain.
Correspondence: Elena Tena Edo (Faculty of Health Sciences, International University of La Rioja, Av. de la Paz, 137, 26006 Logrono, La Rioja, Spain; Email: elenatenaedo@gmail.com).
Annals of Urologic Oncology 2025, 8(2): 76-87. https://doi.org/10.32948/auo.2025.06.02
Received: 15 May 2025 | Accepted: 18 Jul 2025 | Published online: 07 Aug 2025
Key words kidney cancer, ccRCC, VHL, metabolic reprogramming, therapeutics
At the heart of kidney cancer's metabolic transformation lies the constitutive activation of hypoxia-inducible factors (HIFs), particularly HIF-2α, due to VHL loss [6]. Under normal oxygen conditions, VHL targets HIF-α subunits for proteasomal degradation, but in ccRCC, this regulatory mechanism fails, creating a state of pseudohypoxia regardless of actual oxygen availability [7]. HIF stabilization orchestrates a broad transcriptional program that upregulates glucose transporters (GLUT1, GLUT3) and glycolytic enzymes (HK2, PKM2, LDHA), while simultaneously suppressing mitochondrial oxidative phosphorylation [8] [9]. This metabolic shift, reminiscent of the Warburg effect but with unique kidney cancer-specific features, provides rapidly dividing tumor cells with essential biosynthetic precursors while maintaining redox homeostasis [10]. However, recent research has revealed that kidney cancer metabolism extends far beyond glycolysis, encompassing profound alterations in lipid, amino acid, and nucleotide metabolism that collectively sustain tumor growth and survival.
The lipid-rich phenotype of ccRCC represents one of its most distinctive metabolic features, visible histologically as cytoplasmic lipid droplets [11]. This characteristic results from coordinated increases in fatty acid uptake (mediated by CD36 and other transporters), enhanced de novo lipogenesis (through upregulation of FASN and ACC), and impaired lipid oxidation due to mitochondrial dysfunction [12] [13]. The metabolic implications of this lipid reprogramming are multifaceted, providing energy storage, membrane components for rapidly dividing cells, and precursors for signaling molecules that influence tumor progression. Similarly, kidney cancers develop a pronounced dependence on glutamine metabolism, utilizing this amino acid not only as a nitrogen donor for nucleotide synthesis but also as a carbon source for anaplerotic replenishment of TCA cycle intermediates [14] [15]. This metabolic flexibility enables tumors to adapt to nutrient-poor conditions and resist therapeutic interventions.
Beyond cancer cell-intrinsic metabolic changes, kidney tumors actively remodel their microenvironment through metabolic interactions that influence disease progression and treatment response [16]. The glycolytic TME becomes enriched in lactate and other metabolites that suppress immune cell function while promoting angiogenesis [17, 18]. Cancer-associated fibroblasts (CAFs) contribute to this metabolic symbiosis by providing alternative nutrient sources, while endothelial cells adapt to the hypoxic conditions by altering their own metabolic preferences [19, 20]. These complex interactions create therapeutic challenges but also reveal new vulnerabilities that could be exploited for more effective treatments.
The clinical implications of kidney cancer metabolism have become increasingly apparent with the development of targeted therapies. The recent FDA approval of belzutifan, a HIF-2α inhibitor, validates the therapeutic potential of targeting cancer metabolism, while numerous other metabolic inhibitors are in clinical development [21]. However, significant challenges remain, including metabolic heterogeneity within tumors, the development of resistance mechanisms, and the need for reliable biomarkers to guide therapy selection. Emerging technologies such as metabolomic profiling, hyperpolarized MRI, and single-cell analysis are providing unprecedented insights into kidney cancer metabolism, enabling more precise targeting of metabolic vulnerabilities [22].
Lipid metabolism is also profoundly altered, with increased fatty acid uptake and storage to support membrane biosynthesis and energy reserves. Glutamine metabolism is similarly reconfigured, supplying critical precursors for nucleotide synthesis and glutathione production, thereby sustaining proliferation and redox balance [25]. Additionally, mitochondrial dysfunction impairs oxidative phosphorylation, further shifting dependence toward anaerobic metabolic pathways [26]. These adaptations not only fuel tumor growth and survival but also expose metabolic vulnerabilities that could be therapeutically targeted [27]. Elucidating these dysregulated pathways is essential for designing precision therapies to disrupt cancer metabolic dependencies and improve clinical outcomes.
Glycolysis and the warburg effect in kidney cancer
A hallmark of metabolic reprogramming in kidney cancer, particularly ccRCC, is the preferential utilization of glycolysis for energy production even in the presence of oxygen - a phenomenon termed the Warburg effect [6]. This metabolic shift is driven primarily by the constitutive stabilization of HIF-1α and HIF-2α due to loss of the VHL tumor suppressor. The enhanced glycolytic flux provides rapidly proliferating tumor cells with essential biosynthetic intermediates, including nucleotides, amino acids, and lipids, while simultaneously maintaining redox homeostasis through lactate production. Importantly, the Warburg effect supports tumor growth in the typically hypoxic microenvironment of renal carcinomas by reducing oxygen dependence for ATP generation [28]. This metabolic adaptation not only facilitates energy production but also creates a microenvironment that promotes immune evasion and therapeutic resistance [29]. The molecular underpinnings of glycolytic dysregulation in kidney cancer present promising targets for therapeutic intervention, including inhibitors of key glycolytic enzymes and HIF signaling pathways [30, 31].
Lipid metabolism reprogramming in kidney cancer
Kidney cancer exhibits profound alterations in lipid metabolism that support tumor growth and survival [32]. ccRCC is the most common renal malignancy, is particularly characterized by excessive lipid accumulation, visible histologically as cytoplasmic lipid droplets [33]. This metabolic rewiring is driven by multiple mechanisms, including HIF-mediated upregulation of lipid uptake receptors (e.g., CD36), enhanced de novo lipogenesis through increased expression of fatty acid synthase (FASN) and ATP-citrate lyase (ACLY), and impaired lipid oxidation due to mitochondrial dysfunction [13]. The resulting lipid-rich environment not only provides energy stores and membrane building blocks for rapidly proliferating tumor cells but also generates signaling molecules that promote tumor progression [34]. Notably, lipid droplets serve as reservoirs for cholesterol esters and phospholipids that can be mobilized to fuel cancer cell growth under nutrient-deprived conditions [35]. Furthermore, lipid-derived metabolites function as signaling molecules that modulate oncogenic pathways and contribute to the immunosuppressive TME. These metabolic adaptations present promising therapeutic targets, with several inhibitors of lipid metabolism currently under investigation for kidney cancer treatment.
Glutamine dependency and amino acid metabolism in kidney cancer
Renal cell carcinomas, particularly clear cell subtypes, demonstrate marked glutamine addiction as part of their metabolic reprogramming. This dependence stems from the tumor's need to replenish tricarboxylic acid (TCA) cycle intermediates (anaplerosis) and generate biosynthetic precursors for nucleotides, proteins, and antioxidants [36] [37]. The frequent loss of VHL and subsequent HIF stabilization upregulate glutamine transporters (ASCT2, SN2) and key enzymes like glutaminase (GLS), which converts glutamine to glutamate [38]. This metabolic adaptation becomes crucial in kidney cancer as mitochondrial dysfunction limits glucose-derived acetyl-CoA entry into the TCA cycle [6]. Beyond energy production, glutamine metabolism supports redox balance by maintaining glutathione levels and provides nitrogen for non-essential amino acid synthesis through transamination reactions. Interestingly, kidney tumors also alter other amino acid pathways - notably upregulating serine/glycine metabolism for one-carbon units and modulating branched-chain amino acid catabolism. These interconnected amino acid fluxes create metabolic vulnerabilities, with preclinical studies showing sensitivity to glutaminase inhibitors and amino acid deprivation strategies. The emerging understanding of kidney cancer's amino acid metabolic network offers promising therapeutic avenues to target this nutrient dependency while potentially overcoming resistance to conventional therapies [31, 39].
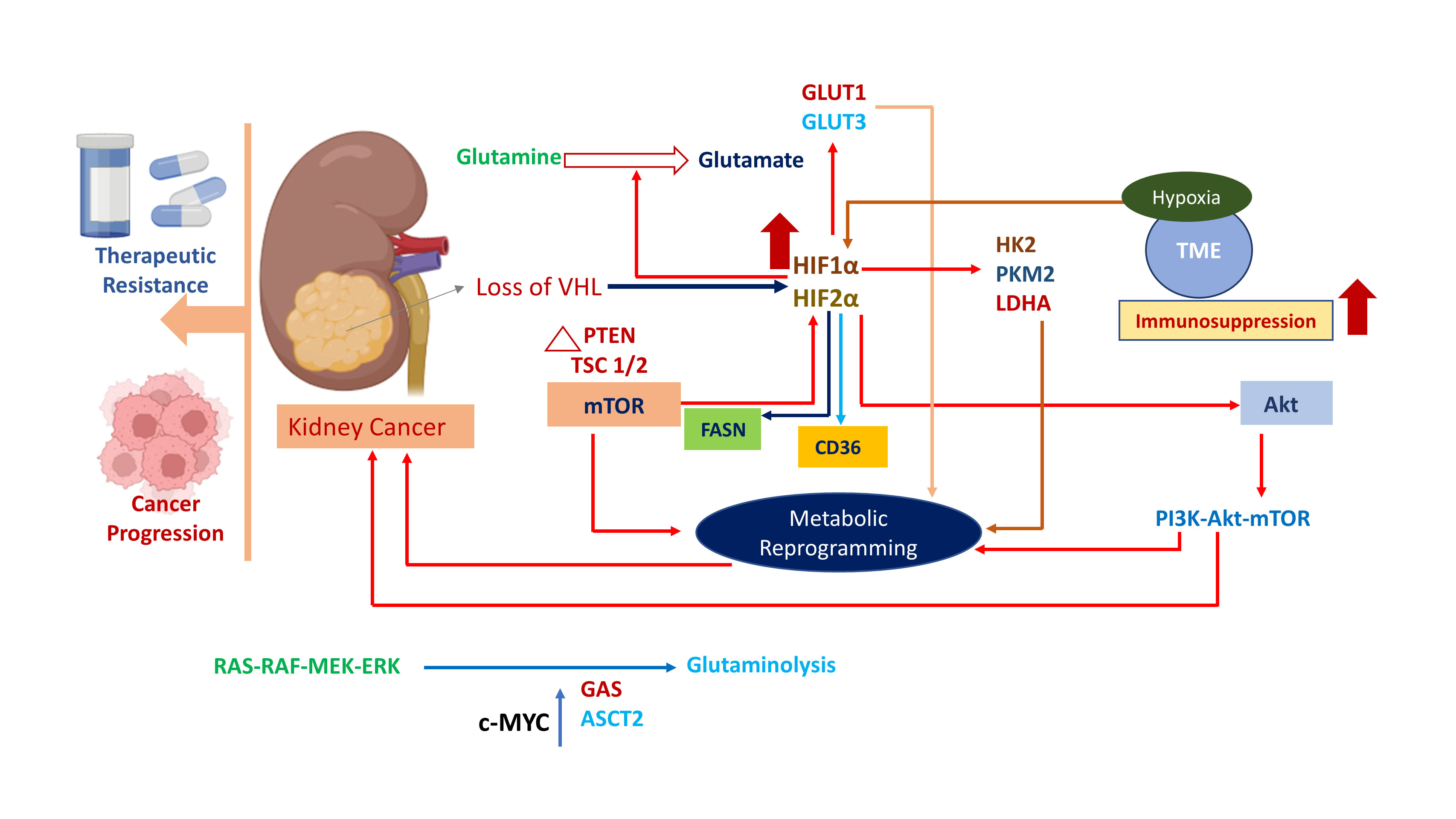 Figure 1. Metabolic reprogramming drives kidney cancer progression. Stabilization and upregulation of HIF1α and HIF2α occur because of Loss of VHL. Metabolic pathways are dysregulated and rewired by alteration of several mediators (GLUT1, GLUT3, HK2, LDHA, etc.) which are activated by the action of HIF1α and HIF2α. Hypoxia is initiated in TME leading to immunosuppression through suppression of cytotoxic T-cells.
Figure 1. Metabolic reprogramming drives kidney cancer progression. Stabilization and upregulation of HIF1α and HIF2α occur because of Loss of VHL. Metabolic pathways are dysregulated and rewired by alteration of several mediators (GLUT1, GLUT3, HK2, LDHA, etc.) which are activated by the action of HIF1α and HIF2α. Hypoxia is initiated in TME leading to immunosuppression through suppression of cytotoxic T-cells.
Hypoxia-inducible factors and their role in kidney cancer pathogenesis
HIF-1α and HIF-2α serve as master regulators of metabolic adaptation in kidney cancer, with their aberrant activation representing a molecular hallmark of ccRCC [44]. The constitutive stabilization of HIF isoforms, primarily resulting from biallelic inactivation of the VHL tumor suppressor, orchestrates a comprehensive transcriptional program that drives tumor progression [45]. HIF activation mediates a pseudo-hypoxic state even under normoxic conditions, upregulating glycolytic enzymes (HK2, LDHA), glucose transporters (GLUT1/3), and angiogenic factors (VEGF) to promote anaerobic metabolism and vascularization [46]. Notably, HIF-2α demonstrates particular oncogenic specificity in ccRCC, enhancing cell proliferation through cyclin D1 regulation while suppressing oxidative phosphorylation [47]. The HIF-mediated metabolic shift also extends to glutaminolysis and lipid storage, creating a tumor-permissive microenvironment [48]. Paradoxically, while HIF-1α often exhibits tumor-suppressive properties in other cancers, both isoforms collaborate in ccRCC to establish the characteristic metabolic phenotype [49]. This unique dependency on HIF signaling presents therapeutic opportunities, with several HIF-2α-specific inhibitors now in clinical development, offering targeted approaches to disrupt the metabolic foundation of kidney cancer.
Mutations in VHL, mTOR, and other metabolic regulators in kidney cancer pathogenesis
The metabolic landscape of kidney cancer is fundamentally shaped by genetic alterations in key regulatory genes, with VHL inactivation representing the seminal event in ccRCC pathogenesis [50]. Biallelic VHL loss triggers constitutive HIF stabilization, establishing the characteristic pseudohypoxic phenotype that drives glycolytic flux and angiogenesis. Complementing this, frequent mutations in mTOR pathway components (e.g., PTEN, TSC1/2) and chromatin remodelers (PBRM1, SETD2, BAP1) create a permissive environment for metabolic reprogramming [51]. The PI3K-AKT-mTOR axis emerges as a critical co-regulator, integrating nutrient availability with biosynthetic demands through control of glycolysis, lipogenesis, and protein synthesis [52]. Notably, these genetic events exhibit functional crosstalk - VHL-deficient cells show heightened mTORC1 sensitivity to amino acids, while epigenetic modifiers influence HIF-target gene accessibility [53]. Additional metabolic regulators like FH and SDH, though less frequently mutated in ccRCC, further demonstrate how mitochondrial dysfunction can propagate oncogenic metabolic shifts [54]. This interconnected mutational architecture not only sustains tumor proliferation but also creates discrete therapeutic vulnerabilities, with current strategies targeting both HIF-dependent (e.g., belzutifan) and mTOR-driven (e.g., everolimus) metabolic pathways [55]. The convergence of these genetic alterations establishes a metabolic framework where nutrient sensing, epigenetic regulation, and oxygen response systems collectively fuel kidney cancer progression.
Oncogenic signaling pathways influencing metabolism in kidney cancer
Kidney cancer pathogenesis is driven by the interplay of multiple oncogenic signaling pathways that collectively reprogram cellular metabolism to support tumor growth and survival [56]. The PI3K-AKT-mTOR axis serves as a central metabolic rheostat, coordinating nutrient uptake and anabolic processes by upregulating glucose transporters (GLUT1/3), glycolytic enzymes (HK2, PKM2), and lipogenic factors (SREBP1, ACLY) [57, 58]. This pathway functionally intersects with HIF signaling - amplified in VHL-deficient tumors - to enhance glycolytic flux while suppressing mitochondrial oxidative phosphorylation [59]. Concurrently, RAS-RAF-MEK-ERK signaling promotes glutaminolysis through c-MYC-mediated upregulation of glutaminase (GAS) and ASCT2 transporters, sustaining TCA cycle anaplerosis [60, 61]. Notably, these pathways exhibit reciprocal regulation: mTORC1 activation stabilizes HIF-α proteins, while HIF-2α transcriptionally activates AKT, creating a feed-forward loop that amplifies metabolic reprogramming [62]. The tumor suppressor p53's frequent inactivation further exacerbates this metabolic shift by relieving repression of glycolysis and disabling oxidative metabolism checkpoints [63]. These interconnected pathways create a permissive metabolic environment characterized by heightened glucose and glutamine dependency, lipid droplet accumulation, and redox adaptation - all exploitable therapeutic vulnerabilities [64, 65]. Current targeted therapies (e.g., mTOR inhibitors, HIF-2α antagonists) and emerging metabolic approaches aim to disrupt these oncogenic signaling-metabolism nexuses in kidney cancer.
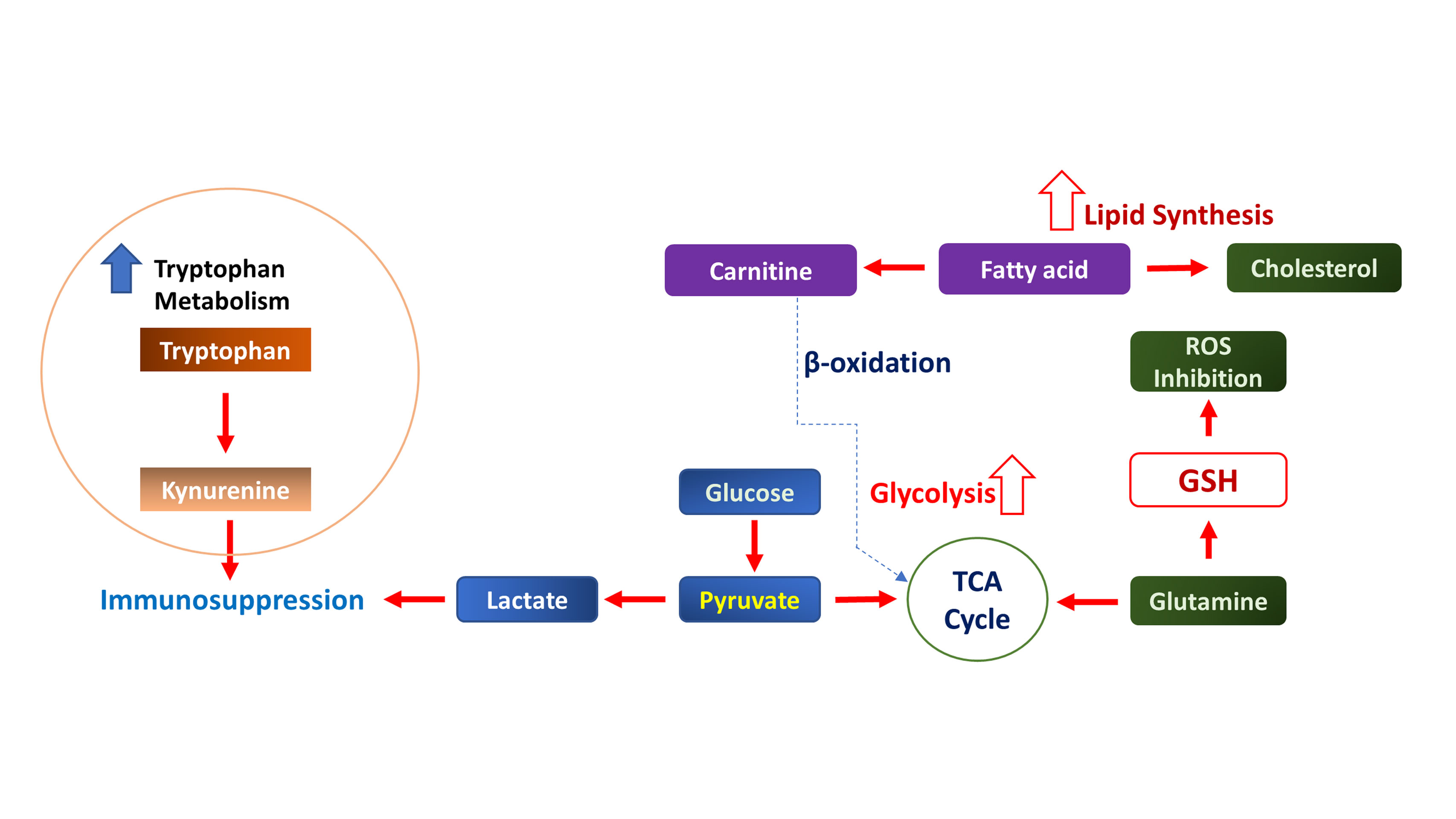 Figure 2. Metabolic reprogramming and immunosuppressive networks in kidney cancer. Enhanced glycolysis converts glucose to lactate, acidifying the TME and promoting immunosuppression. TCA cycle disruption drives oncogenic signaling in kidney cancer. Fatty acid synthesis and cholesterol accumulation sustain membrane biogenesis and signaling. Glutathione (GSH) synthesis neutralizes ROS, enabling chemoresistance.
Figure 2. Metabolic reprogramming and immunosuppressive networks in kidney cancer. Enhanced glycolysis converts glucose to lactate, acidifying the TME and promoting immunosuppression. TCA cycle disruption drives oncogenic signaling in kidney cancer. Fatty acid synthesis and cholesterol accumulation sustain membrane biogenesis and signaling. Glutathione (GSH) synthesis neutralizes ROS, enabling chemoresistance.
Crosstalk between tumor cells and stroma in kidney cancer
The bidirectional metabolic interplay between tumor cells and stromal components in kidney cancer creates a dynamic microenvironment that fuels disease progression. CAFs actively secrete lactate, pyruvate, and ketone bodies that tumor cells utilize as alternative energy substrates through oxidative phosphorylation, particularly under glucose-deprived conditions [69]. Conversely, tumor cells release glutamate and other oncometabolites that activate CAFs, inducing their transformation into myofibroblasts that further remodel the extracellular matrix [73]. This metabolic symbiosis extends to endothelial cells, where HIF-driven VEGF secretion from tumor cells promotes angiogenesis, while the resulting neovasculature provides nutrients and oxygen that sustain tumor growth [74]. Adipocytes in perirenal fat deposits contribute free fatty acids that tumor cells internalize through CD36-mediated uptake, supporting membrane biosynthesis and energy storage [75]. Importantly, this crosstalk is mediated by exosomal transfer of miRNAs and metabolic enzymes that reprogram recipient cells. The resulting metabolic coupling not only enhances tumor survival under stress conditions but also creates therapeutic resistance by establishing redundant nutrient acquisition pathways. Targeting these tumor-stroma metabolic interactions – through approaches like CAF depletion, anti-angiogenic therapy, or lipid metabolism inhibition – represents a promising strategy to disrupt the tumor-supportive niche in kidney cancer.
Immune cell metabolism and immunosuppression in the kidney cancer microenvironment
The metabolic landscape of kidney cancer actively shapes antitumor immunity by imposing nutrient constraints and altering immune cell functionality within the TME. Tumor cells outcompete infiltrating lymphocytes for glucose through elevated expression of GLUT1 and hexokinase-2, forcing cytotoxic T cells into a hypofunctional state characterized by impaired glycolysis and reduced interferon-γ production [76]. Conversely, myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) thrive in this metabolically hostile environment by preferentially utilizing fatty acid oxidation and oxidative phosphorylation, which support their immunosuppressive functions [77]. The accumulation of tumor-derived lactate and kynurenine further reinforces immunosuppression by inhibiting natural killer cell activity while promoting the polarization of tumor-associated macrophages toward an M2 phenotype. Notably, the hypoxic tumor core drives PD-L1 upregulation on both cancer cells and infiltrating myeloid cells through HIF-1α stabilization, creating an immune checkpoint-rich environment (Figure 3) [78]. These metabolic constraints contribute to the limited efficacy of immunotherapies in kidney cancer, prompting investigations into metabolic modulators – such as lactate dehydrogenase inhibitors and IDO1 antagonists – that may reverse immunosuppression and enhance checkpoint blockade responses [79]. Understanding these immunometabolic interactions provides critical insights for developing combination strategies that simultaneously target tumor metabolism and immune evasion mechanisms.
Angiogenesis and nutrient supply in kidney cancer progression
Kidney cancer orchestrates a robust angiogenic response to sustain its metabolic demands through complex interactions between tumor cells and the vascular microenvironment. The characteristic VHL/HIF axis activation in ccRCC drives excessive vascular endothelial growth factor (VEGF) production [53], stimulating the formation of disorganized, hyperpermeable tumor vasculature. These aberrant vessels, while providing increased nutrient and oxygen supply, create a paradoxical state of chronic hypoxia due to their structural abnormalities and inefficient perfusion. Tumor cells adapt by further upregulating HIF-dependent glycolytic enzymes and glucose transporters, establishing a self-perpetuating cycle of metabolic demand and vascular recruitment [80, 81]. The resulting vasculature not only delivers glucose and glutamine but also serves as a conduit for lipid uptake from circulating lipoproteins, supporting the lipid droplet accumulation characteristic of ccRCC. Importantly, the angiogenic switch enables metastatic dissemination by providing tumor cells access to systemic circulation while simultaneously creating an immunosuppressive microenvironment through VEGF-mediated inhibition of dendritic cell maturation [82]. This understanding has led to the clinical success of anti-angiogenic therapies, though their efficacy is often limited by the emergence of alternative nutrient acquisition strategies, including enhanced macropinocytosis and vascular co-option. Current research focuses on combining VEGF pathway inhibitors with metabolic or immunotherapeutic agents to more effectively starve tumors while preventing compensatory adaptations.
 Figure 3. Immunosuppressive molecular mechanism in kidney cancer. LDH facilitates conversion of tumor-derived Lactate from pyruvate. The accumulation of LDH acidifies the TME leading to inhibition of cytotoxic T-cell functions, NK cell functions, and modulation of tumor associated macrophages (TAM). As a result, Treg cells are activated which further promotes cancer cell progression.
Figure 3. Immunosuppressive molecular mechanism in kidney cancer. LDH facilitates conversion of tumor-derived Lactate from pyruvate. The accumulation of LDH acidifies the TME leading to inhibition of cytotoxic T-cell functions, NK cell functions, and modulation of tumor associated macrophages (TAM). As a result, Treg cells are activated which further promotes cancer cell progression.
Metabolic biomarkers in kidney cancer: current applications and emerging potential
The unique metabolic rewiring of kidney cancer has yielded clinically relevant biomarkers that enhance diagnostic precision, prognostic stratification, and therapeutic monitoring. ccRCC-specific metabolic signatures—including elevated circulating succinate levels from pseudohypoxic drive and increased urinary N-acetylaspartate reflecting altered lipid metabolism—provide non-invasive diagnostic indicators that complement imaging findings [87]. Prognostically, immunohistochemical detection of key metabolic enzymes (CAIX, GLUT1) in tumor tissues stratifies patient risk, while liquid biopsy profiles measuring kynurenine/tryptophan ratios or branched-chain amino acid patterns predict immunotherapy response [88]. Advanced imaging biomarkers, particularly 18F-FDG PET avidity and hyperpolarized 13C-pyruvate MRI, quantitatively map tumor glycolytic activity, correlating with tumor grade and metastatic potential [89]. Emerging mass spectrometry-based metabolomics now identify signature perturbations in TCA cycle intermediates (fumarate, 2-HG) that reveal underlying genetic alterations (FH/SDH mutations) and guide targeted therapy selection [90]. Notably, dynamic changes in serum acylcarnitine profiles and extracellular vesicle-derived metabolic enzymes show promise for real-time treatment monitoring. These biomarkers collectively address critical clinical challenges in kidney cancer management, from differentiating indolent from aggressive disease to detecting micro-metastases and overcoming therapeutic resistance. Their integration into multi-omics diagnostic platforms is advancing personalized management strategies that align tumor-specific metabolic vulnerabilities with precision therapies.
Imaging techniques for metabolic profiling in kidney cancer
Advanced imaging modalities now enable non-invasive metabolic profiling of kidney tumors, providing critical diagnostic and prognostic information while guiding treatment decisions. Positron emission tomography (PET) using 18F-fluorodeoxyglucose (FDG) remains the cornerstone for evaluating glycolytic activity, with standardized uptake values (SUVmax) correlating with tumor aggressiveness and metastatic potential [91, 92]. Novel PET tracers targeting other metabolic pathways—such as 11C-acetate for lipid metabolism and 18F-fluoroglutamine for amino acid uptake—are expanding the metabolic profiling capabilities [93]. Magnetic resonance spectroscopy (MRS) offers complementary data by quantifying endogenous metabolites, including elevated choline peaks reflecting membrane turnover and reduced citrate levels characteristic of ccRCC. Emerging hyperpolarized 13C-pyruvate MRI techniques dynamically track real-time conversion of pyruvate to lactate, directly visualizing Warburg effect activity with unprecedented spatial resolution [94]. Chemical shift imaging reliably detects intracellular lipid content, distinguishing clear cell from non-clear cell variants with >90% accuracy [95]. These functional imaging approaches are being integrated with radiomic analysis of conventional CT/MRI to create multiparametric metabolic signatures that predict treatment response and monitor therapeutic efficacy. The non-invasive nature of metabolic imaging positions it as an ideal tool for serial assessment during therapy, particularly for evaluating emerging metabolism-targeted treatments like HIF-2α inhibitors and glutaminase blockers.
Inhibitors of glycolysis and HIF signaling in kidney cancer therapeutics
The targeting of glycolytic and HIF signaling pathways represents a precision medicine approach for kidney cancer, capitalizing on the tumor's hallmark metabolic vulnerabilities. HIF-2α antagonists such as belzutifan (MK-6482) have demonstrated clinical efficacy by specifically disrupting the pseudohypoxic transcriptional program in VHL-deficient tumors, reducing expression of glycolytic enzymes (HK2, LDHA) and glucose transporters (GLUT1/3) [107]. Parallel strategies employ small molecule inhibitors of rate-limiting glycolytic components—including 2-deoxyglucose (glycolytic inhibitor) and PFK158 (PFKFB3 blocker)—to starve tumors of their preferred energy source while sparing normal cells that retain oxidative phosphorylation capacity [108]. Particularly promising are dual-action compounds that concurrently target HIF signaling and glycolysis, such as PT2385 derivatives that destabilize HIF-2α while inhibiting hexokinase activity [109]. These approaches show synergistic potential when combined with anti-angiogenic therapies, as HIF inhibition normalizes tumor vasculature while glycolytic blockade prevents metabolic adaptation. Resistance mechanisms, including upregulation of alternate HIF isoforms or activation of compensatory nutrient salvage pathways, are being addressed through next-generation inhibitors with improved target specificity and combination regimens incorporating glutaminase blockers. The development of PET-based biomarkers (18F-FDG, 18F-fluoromisonidazole) enables real-time monitoring of therapeutic response, facilitating dose optimization for these metabolism-targeted agents [110]. This therapeutic paradigm exemplifies how understanding cancer-specific metabolic dependencies can yield targeted treatments with potentially fewer off-target effects than conventional therapies.
Targeting lipid and amino acid metabolism in kidney cancer therapy
Emerging therapeutic strategies are exploiting the deregulated lipid and amino acid metabolism that underlies kidney cancer pathogenesis. The characteristic lipid droplet accumulation in clear cell RCC has prompted development of fatty acid synthase (FASN) inhibitors like TVB-2640, which disrupt de novo lipogenesis and induce tumor-specific apoptosis by depriving cancer cells of membrane precursors and signaling lipids [111]. Concurrently, inhibitors of sterol regulatory element-binding proteins (SREBP) such as fatostatin are being evaluated to block the lipogenic transcription program driven by HIF and mTOR pathways [112]. In amino acid metabolism, glutaminase inhibitors (telaglenastat) and ASCT2 blockers (V-9302) are showing promise in clinical trials by restricting tumor access to glutamine – a crucial nitrogen and carbon source for ccRCC proliferation [113]. Notably, these approaches synergize with existing therapies: lipid metabolism inhibitors enhance anti-angiogenic efficacy by reducing VEGF production, while amino acid restriction potentiates immunotherapy by alleviating immunosuppressive tryptophan/kynurenine pathways. Advanced patient stratification using lipidomic profiles and PET imaging with glutamine analogs (18F-FGln) is enabling precision targeting of these metabolic vulnerabilities. The simultaneous targeting of both lipid and amino acid pathways represents a multipronged strategy to overwhelm tumor metabolic plasticity and overcome treatment resistance in kidney cancer.
Combination therapies: metabolic drugs with immunotherapy/TKI in kidney cancer treatment
The strategic integration of metabolic modulators with immunotherapy and tyrosine kinase inhibitors (TKIs) represents a paradigm shift in kidney cancer treatment, addressing both tumor-intrinsic vulnerabilities and microenvironmental immunosuppression. Preclinical studies demonstrate that HIF-2α inhibitors (belzutifan) synergize with PD-1/PD-L1 blockade by alleviating hypoxia-driven immunosuppression while normalizing aberrant tumor vasculature when combined with VEGF-targeted TKIs [114]. Clinically, glutaminase inhibitors (telaglenastat) are being evaluated with pembrolizumab to simultaneously restrict tumor bioenergetics and enhance T-cell function by reducing myeloid-derived suppressor cell (MDSC) accumulation in the TME [115]. Similarly, lactate dehydrogenase inhibitors (GSK2837808A) are showing promise in combination regimens by reversing the lactate-mediated suppression of cytotoxic T lymphocytes while maintaining anti-angiogenic effects of TKIs [116]. Emerging trial data reveal that these combinations yield durable responses by targeting complementary resistance mechanisms—metabolic drugs prevent the glycolytic adaptation that often limits TKI efficacy, while immunotherapy counters the immunosuppressive effects of metabolic stress. Advanced biomarker strategies, including metabolic PET imaging (18F-FDG, 18F-FSPG) and immune-metabolic profiling of tumor biopsies, are enabling real-time monitoring of these synergistic effects [117]. This multidimensional therapeutic approach capitalizes on the interconnected nature of metabolic and signaling networks in kidney cancer, offering new avenues to overcome treatment resistance and improve long-term outcomes.
|
Table 1. Anticancer drugs effective in kidney cancer. |
|||
|
Name of the Drug |
Manufacturer |
Mechanism |
Reference |
|
Lonidamine |
Angelini Pharma |
Hexokinase-2 inhbitor |
[102] |
|
Belzutifan |
Merck & Co. |
Targeting HIF-2α |
[103] |
|
Telaglenastat |
Calithera Biosciences |
Glutamine pathway inhibitor |
[104] |
|
TVB-2640 |
Sagimet Biosciences |
FASN inhibitor |
[105] |
|
FX11 |
Albert Einstein college of Medicine |
LDHA inhibitor |
[106] |
Resistance to metabolic therapies in kidney cancer
The emergence of resistance to metabolism-targeted agents in kidney cancer stems from the remarkable metabolic plasticity and genetic adaptability of tumor cells. A primary mechanism involves compensatory upregulation of alternative nutrient acquisition pathways—for instance, tumors treated with glutaminase inhibitors frequently activate macropinocytosis to scavenge extracellular proteins or amplify ASCT2-independent glutamine transport systems [121]. Similarly, glycolytic blockade often triggers a metabolic shift toward oxidative phosphorylation through mitochondrial genome amplification or increased fatty acid β-oxidation [122]. Epigenetic remodeling enables rapid adaptation, with demethylation of metabolic gene promoters facilitating expression of bypass pathways under therapeutic pressure. The TME further contributes to resistance through metabolic symbiosis, where stromal cells supply metabolites (lactate, ketones) that rescue treated tumor cells from energy crisis. Heterogeneous expression of metabolic enzymes across tumor subclones creates inherent resistance reservoirs, while HIF stabilization in perinecrotic regions maintains tumor survival despite therapy [123]. Emerging strategies to overcome resistance include intermittent dosing to prevent adaptive responses, dual targeting of complementary metabolic nodes (e.g., concurrent glycolysis and OXPHOS inhibition), and combining metabolic drugs with epigenetic modifiers to limit transcriptional adaptation. The development of functional metabolic imaging techniques (hyperpolarized MRI, metabolic PET tracers) now enables real-time monitoring of these resistance mechanisms, guiding adaptive therapeutic strategies in clinical trials.
Emerging technologies and novel therapeutic targets in kidney cancer metabolism
Recent advances in multi-omics technologies and high-resolution metabolic imaging are uncovering novel therapeutic vulnerabilities in kidney cancer metabolism. Single-cell metabolomics has revealed previously unappreciated metabolic heterogeneity within tumors, identifying rare subpopulations with dependencies on cysteine or one-carbon metabolism that could be targeted with new small-molecule inhibitors [124]. CRISPR-based metabolic gene screening has pinpointed hexosamine biosynthesis and serine/glycine conversion as essential pathways in VHL-deficient cells, while spatial transcriptomics maps metabolic crosstalk between tumor and immune cells within the TME [125]. Emerging therapeutic targets include the mitochondrial pyruvate carrier (MPC) – inhibition of which selectively starves kidney cancer cells of TCA cycle intermediates – and the cystine/glutamate antiporter xCT, which maintains redox balance in metastatic lesions. Nanotechnology approaches are enabling targeted delivery of metabolic drugs, such as nanoparticle-encapsulated glutaminase inhibitors that preferentially accumulate in tumors. Meanwhile, AI-driven analysis of metabolic flux data is predicting patient-specific vulnerabilities by modeling individual tumor metabolic networks. These innovations are being translated clinically through innovative trial designs, including basket trials testing metabolic therapies based on molecular features rather than histology, and window-of-opportunity studies using hyperpolarized 13C-MRI to quantify real-time drug effects on tumor metabolism [101]. Together, these technological advances are expanding the arsenal of metabolism-targeted therapies while enabling precision approaches tailored to individual patient tumors.
None.
Ethical policy
Non applicable.
Availability of data and materials
All data generated or analysed during this study are included in this publication.
Author contributions
Elena Tena Edo contributed to design of the work, data collection, and drafting the article.
Competing interests
The author declares no competing interests.
Funding
None.
- Hu J, Wang S-G, Hou Y, Chen Z, Liu L, Li R, Li N, Zhou L, Yang Y, Wang L et al: Multi-omic profiling of clear cell renal cell carcinoma identifies metabolic reprogramming associated with disease progression. Nature Genetics 2024, 56(3): 442-457.
- Sudarshan S, Karam JA, Brugarolas J, Thompson RH, Uzzo R, Rini B, Margulis V, Patard JJ, Escudier B, Linehan WM: Metabolism of kidney cancer: from the lab to clinical practice. Eur Urol 2013, 63(2): 244-251.
- Carthew RW: Gene Regulation and Cellular Metabolism: An Essential Partnership. Trends Genet 2021, 37(4): 389-400.
- Liu Y, Zhao Y, Song H, Li Y, Liu Z, Ye Z, Zhao J, Wu Y, Tang J, Yao M: Metabolic reprogramming in tumor immune microenvironment: Impact on immune cell function and therapeutic implications. Cancer Letters 2024, 597: 217076.
- Mao Y, Xia Z, Xia W, Jiang P: Metabolic reprogramming, sensing, and cancer therapy. Cell Reports 2024, 43(12): 115064.
- Chen Z, Zhang X: The role of metabolic reprogramming in kidney cancer. Front Oncol 2024, 14: 1402351.
- Haase VH: The VHL/HIF oxygen-sensing pathway and its relevance to kidney disease. Kidney International 2006, 69(8): 1302-1307.
- Zhang J, Yao M, Xia S, Zeng F, Liu Q: Systematic and comprehensive insights into HIF-1 stabilization under normoxic conditions: implications for cellular adaptation and therapeutic strategies in cancer. Cellular & Molecular Biology Letters 2025, 30(1): 2.
- Nsiah NY, Morgan AB, Donkor N, Inman DM: Long-term HIF-1α stabilization reduces respiration, promotes mitophagy, and results in retinal cell death. Scientific Reports 2023, 13(1): 20541.
- Mathew M, Nguyen NT, Bhutia YD, Sivaprakasam S, Ganapathy V: Metabolic Signature of Warburg Effect in Cancer: An Effective and Obligatory Interplay between Nutrient Transporters and Catabolic/Anabolic Pathways to Promote Tumor Growth. Cancers (Basel) 2024, 16(3): 504.
- Sanchez DJ, Simon MC: Genetic and metabolic hallmarks of clear cell renal cell carcinoma. Biochim Biophys Acta Rev Cancer 2018, 1870(1): 23-31.
- Wang J, Guo H, Zheng L-F, Li P, Zhao T-J: Context-specific fatty acid uptake is a finely-tuned multi-level effort. Trends in Endocrinology & Metabolism 2024, https://doi.org/https://doi.org/10.1016/j.tem.2024.10.001. Epub ahead of print.
- Schroeder B, Vander Steen T, Espinoza I, Venkatapoorna CMK, Hu Z, Silva FM, Regan K, Cuyàs E, Meng XW, Verdura S et al: Fatty acid synthase (FASN) regulates the mitochondrial priming of cancer cells. Cell Death & Disease 2021, 12(11): 977.
- Abu Aboud O, Habib SL, Trott J, Stewart B, Liang S, Chaudhari AJ, Sutcliffe J, Weiss RH: Glutamine Addiction in Kidney Cancer Suppresses Oxidative Stress and Can Be Exploited for Real-Time Imaging. Cancer Res 2017, 77(23): 6746-6758.
- Jin J, Byun J-K, Choi Y-K, Park K-G: Targeting glutamine metabolism as a therapeutic strategy for cancer. Experimental & Molecular Medicine 2023, 55(4): 706-715.
- Elia I, Haigis MC: Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nat Metab 2021, 3(1): 21-32.
- Wang Z-H, Peng W-B, Zhang P, Yang X-P, Zhou Q: Lactate in the tumour microenvironment: From immune modulation to therapy. EBioMedicine 2021, 73: 103627.
- Pérez-Tomás R, Pérez-Guillén I: Lactate in the Tumor Microenvironment: An Essential Molecule in Cancer Progression and Treatment. Cancers (Basel) 2020, 12(11): 3244.
- Li Z, Sun C, Qin Z: Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics 2021, 11(17): 8322-8336.
- Xia B, Qiu L, Yue J, Si J, Zhang H: The metabolic crosstalk of cancer-associated fibroblasts and tumor cells: Recent advances and future perspectives. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 2024, 1879(6): 189190.
- Wu X, Lazris D, Wong R, Tykodi SS: Belzutifan for the treatment of renal cell carcinoma. Ther Adv Med Oncol 2025, 17: 17588359251317846.
- Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, Oudard S, Else T, Maranchie JK, Welsh SJ et al: Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N Engl J Med 2021, 385(22): 2036-2046.
- Zheng X, Liu Y, Yang Z, Tian Y: Metabolic reprogramming and immune microenvironment profiling in clear cell renal cell carcinoma: implications for prognosis, targeted therapy, and drug resistance. Discover Oncology 2025, 16(1): 850.
- Kierans SJ, Taylor CT: Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. The Journal of Physiology 2021, 599(1): 23-37.
- Yoo HC, Yu YC, Sung Y, Han JM: Glutamine reliance in cell metabolism. Experimental & Molecular Medicine 2020, 52(9): 1496-1516.
- Bhatti JS, Bhatti GK, Reddy PH: Mitochondrial dysfunction and oxidative stress in metabolic disorders — A step towards mitochondria based therapeutic strategies. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2017, 1863(5): 1066-1077.
- Tufail M, Jiang C-H, Li N: Altered metabolism in cancer: insights into energy pathways and therapeutic targets. Molecular Cancer 2024, 23(1): 203.
- Liberti MV, Locasale JW: The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci 2016, 41(3): 211-218.
- Zhang H, Li S, Wang D, Liu S, Xiao T, Gu W, Yang H, Wang H, Yang M, Chen P: Metabolic reprogramming and immune evasion: the interplay in the tumor microenvironment. Biomarker Research 2024, 12(1): 96.
- Rabelink TJ, Carmeliet P: Glycolytic adaptation and progression of kidney disease. Nature Reviews Nephrology 2018, 14(2): 75-76.
- Rathmell WK, Rathmell JC, Linehan WM: Metabolic Pathways in Kidney Cancer: Current Therapies and Future Directions. J Clin Oncol 2018, https://doi.org/10.1200/jco.2018.79.2309. Epub ahead of print.:Jco2018792309.
- Zhang Y, Zhang S, Sun H, Xu L: The pathogenesis and therapeutic implications of metabolic reprogramming in renal cell carcinoma. Cell Death Discovery 2025, 11(1): 186.
- Qi X, Li Q, Che X, Wang Q, Wu G: The Uniqueness of Clear Cell Renal Cell Carcinoma: Summary of the Process and Abnormality of Glucose Metabolism and Lipid Metabolism in ccRCC. Front Oncol 2021, 11: 727778.
- Corn KC, Windham MA, Rafat M: Lipids in the tumor microenvironment: From cancer progression to treatment. Progress in Lipid Research 2020, 80: 101055.
- Olzmann JA, Carvalho P: Dynamics and functions of lipid droplets. Nature Reviews Molecular Cell Biology 2019, 20(3): 137-155.
- Eniafe J, Jiang S: The functional roles of TCA cycle metabolites in cancer. Oncogene 2021, 40(19): 3351-3363.
- Anderson NM, Mucka P, Kern JG, Feng H: The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9(2): 216-237.
- Haase VH: The VHL tumor suppressor: master regulator of HIF. Curr Pharm Des 2009, 15(33): 3895-3903.
- Do LK, Lee HM, Ha Y-S, Lee C-H, Kim J: Amino acids in cancer: Understanding metabolic plasticity and divergence for better therapeutic approaches. Cell Reports 2025, 44(4): 115529.
- Miller F, Kentsis A, Osman R, Pan Z-Q: Inactivation of VHL by Tumorigenic Mutations That Disrupt Dynamic Coupling of the pVHL·Hypoxia-inducible Transcription Factor-1α Complex. Journal of Biological Chemistry 2005, 280(9): 7985-7996.
- Ni X, Lu C-p, Xu G-q, Ma J-j: Transcriptional regulation and post-translational modifications in the glycolytic pathway for targeted cancer therapy. Acta Pharmacologica Sinica 2024, 45(8): 1533-1555.
- Walton J, Lawson K, Prinos P, Finelli A, Arrowsmith C, Ailles L: PBRM1, SETD2 and BAP1 — the trinity of 3p in clear cell renal cell carcinoma. Nature Reviews Urology 2023, 20(2): 96-115.
- Singleton DC, Macann A, Wilson WR: Therapeutic targeting of the hypoxic tumour microenvironment. Nature Reviews Clinical Oncology 2021, 18(12): 751-772.
- Hoefflin R, Harlander S, Schäfer S, Metzger P, Kuo F, Schönenberger D, Adlesic M, Peighambari A, Seidel P, Chen C-y et al: HIF-1α and HIF-2α differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nature Communications 2020, 11(1): 4111.
- Tanimoto K, Makino Y, Pereira T, Poellinger L: Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. Embo j 2000, 19(16): 4298-4309.
- Hayashi Y, Yokota A, Harada H, Huang G: Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1α in cancer. Cancer Sci 2019, 110(5): 1510-1517.
- Yang Z, Wei X, Ji C, Ren X, Su W, Wang Y, Zhou J, Zhao Z, Zhou P, Zhao K et al: OGT/HIF--2α axis promotes the progression of clear cell renal cell carcinoma and regulates its sensitivity to ferroptosis. iScience 2023, 26(11): 108148.
- Sun RC, Denko NC: Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab 2014, 19(2): 285-292.
- Cowman SJ, Koh MY: Revisiting the HIF switch in the tumor and its immune microenvironment. Trends in Cancer 2022, 8(1): 28-42.
- Shenoy N, Pagliaro L: Sequential pathogenesis of metastatic VHL mutant clear cell renal cell carcinoma: putting it together with a translational perspective. Annals of Oncology 2016, 27(9): 1685-1695.
- Roldan-Romero JM, Beuselinck B, Santos M, Rodriguez-Moreno JF, Lanillos J, Calsina B, Gutierrez A, Tang K, Lainez N, Puente J et al: PTEN expression and mutations in TSC1, TSC2 and MTOR are associated with response to rapalogs in patients with renal cell carcinoma. Int J Cancer 2020, 146(5): 1435-1444.
- Hassan B, Akcakanat A, Holder AM, Meric-Bernstam F: Targeting the PI3-kinase/Akt/mTOR signaling pathway. Surg Oncol Clin N Am 2013, 22(4): 641-664.
- Mazumder S, Higgins PJ, Samarakoon R: Downstream Targets of VHL/HIF-α Signaling in Renal Clear Cell Carcinoma Progression: Mechanisms and Therapeutic Relevance. Cancers (Basel) 2023, 15(4): 1316.
- Yoo A, Tang C, Zucker M, Fitzgerald K, DiNatale RG, Rappold PM, Weiss K, Freeman B, Lee C-H, Schultz N et al: Genomic and Metabolic Hallmarks of SDH- and FH-deficient Renal Cell Carcinomas. European Urology Focus 2022, 8(5): 1278-1288.
- Yuan X, Ruan W, Bobrow B, Carmeliet P, Eltzschig HK: Targeting hypoxia-inducible factors: therapeutic opportunities and challenges. Nature Reviews Drug Discovery 2024, 23(3): 175-200.
- Xie D, Li G, Zheng Z, Zhang X, Wang S, Jiang B, Li X, Wang X, Wu G: The molecular code of kidney cancer: A path of discovery for gene mutation and precision therapy. Molecular Aspects of Medicine 2025, 101: 101335.
- Aoki M, Fujishita T: Oncogenic Roles of the PI3K/AKT/mTOR Axis. Curr Top Microbiol Immunol 2017, 407: 153-189.
- Glaviano A, Foo ASC, Lam HY, Yap KCH, Jacot W, Jones RH, Eng H, Nair MG, Makvandi P, Geoerger B et al: PI3K/AKT/mTOR signaling transduction pathway and targeted therapies in cancer. Mol Cancer 2023, 22(1): 138.
- Kapitsinou PP, Haase VH: The VHL tumor suppressor and HIF: insights from genetic studies in mice. Cell Death & Differentiation 2008, 15(4): 650-659.
- Stefan E, Bister K: MYC and RAF: Key Effectors in Cellular Signaling and Major Drivers in Human Cancer. Curr Top Microbiol Immunol 2017, 407: 117-151.
- Ritt DA, Abreu-Blanco MT, Bindu L, Durrant DE, Zhou M, Specht SI, Stephen AG, Holderfield M, Morrison DK: Inhibition of Ras/Raf/MEK/ERK Pathway Signaling by a Stress-Induced Phospho-Regulatory Circuit. Molecular Cell 2016, 64(5): 875-887.
- Sakamoto T, Weng JS, Hara T, Yoshino S, Kozuka-Hata H, Oyama M, Seiki M: Hypoxia-inducible factor 1 regulation through cross talk between mTOR and MT1-MMP. Mol Cell Biol 2014, 34(1): 30-42.
- Liu J, Zhang C, Hu W, Feng Z: Tumor suppressor p53 and metabolism. J Mol Cell Biol 2019, 11(4): 284-292.
- Tufail M, Jiang CH, Li N: Altered metabolism in cancer: insights into energy pathways and therapeutic targets. Mol Cancer 2024, 23(1): 203.
- Bosch M, Sweet MJ, Parton RG, Pol A: Lipid droplets and the host-pathogen dynamic: FATal attraction? J Cell Biol 2021, 220(8): e202104005.
- Kay EJ, Zanivan S: The tumor microenvironment is an ecosystem sustained by metabolic interactions. Cell Reports 2025, 44(3): 115432.
- Zhou D, Duan Z, Li Z, Ge F, Wei R, Kong L: The significance of glycolysis in tumor progression and its relationship with the tumor microenvironment. Front Pharmacol 2022, 13: 1091779.
- Zhang A, Fan T, Liu Y, Yu G, Li C, Jiang Z: Regulatory T cells in immune checkpoint blockade antitumor therapy. Molecular Cancer 2024, 23(1): 251.
- Avagliano A, Granato G, Ruocco MR, Romano V, Belviso I, Carfora A, Montagnani S, Arcucci A: Metabolic Reprogramming of Cancer Associated Fibroblasts: The Slavery of Stromal Fibroblasts. Biomed Res Int 2018, 2018: 6075403.
- Bakleh MZ, Al Haj Zen A: The Distinct Role of HIF-1α and HIF-2α in Hypoxia and Angiogenesis. Cells 2025, 14(9): 673.
- Sieow JL, Gun SY, Wong SC: The Sweet Surrender: How Myeloid Cell Metabolic Plasticity Shapes the Tumor Microenvironment. Front Cell Dev Biol 2018, 6: 168.
- Fu Z, Mowday AM, Smaill JB, Hermans IF, Patterson AV: Tumour Hypoxia-Mediated Immunosuppression: Mechanisms and Therapeutic Approaches to Improve Cancer Immunotherapy. Cells 2021, 10(5): 1006.
- Zhang F, Ma Y, Li D, Wei J, Chen K, Zhang E, Liu G, Chu X, Liu X, Liu W et al: Cancer associated fibroblasts and metabolic reprogramming: unraveling the intricate crosstalk in tumor evolution. Journal of Hematology & Oncology 2024, 17(1): 80.
- Lee S, Goldfinger LE: RLIP76 regulates HIF-1 activity, VEGF expression and secretion in tumor cells, and secretome transactivation of endothelial cells. Faseb j 2014, 28(9): 4158-4168.
- Pohl J, Ring A, Korkmaz U, Ehehalt R, Stremmel W: FAT/CD36-mediated long-chain fatty acid uptake in adipocytes requires plasma membrane rafts. Mol Biol Cell 2005, 16(1): 24-31.
- Ganjoo S, Gupta P, Corbali HI, Nanez S, Riad TS, Duong LK, Barsoumian HB, Masrorpour F, Jiang H, Welsh JW et al: The role of tumor metabolism in modulating T-Cell activity and in optimizing immunotherapy. Front Immunol 2023, 14: 1172931.
- Wang H, Zhou F, Qin W, Yang Y, Li X, Liu R: Metabolic regulation of myeloid-derived suppressor cells in tumor immune microenvironment: targets and therapeutic strategies. Theranostics 2025, 15(6): 2159-2184.
- Bailey CM, Liu Y, Liu M, Du X, Devenport M, Zheng P, Liu Y, Wang Y: Targeting HIF-1α abrogates PD-L1-mediated immune evasion in tumor microenvironment but promotes tolerance in normal tissues. J Clin Invest 2022, 132(9): e150846.
- Li H, Zhao A, Li M, Shi L, Han Q, Hou Z: Targeting T-cell metabolism to boost immune checkpoint inhibitor therapy. Front Immunol 2022, 13: 1046755.
- Kierans SJ, Taylor CT: Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol 2021, 599(1): 23-37.
- Schiliro C, Firestein BL: Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10(5): 1056.
- Fricke I, Mirza N, Dupont J, Lockhart C, Jackson A, Lee J-H, Sosman JA, Gabrilovich DI: Vascular Endothelial Growth Factor-Trap Overcomes Defects in Dendritic Cell Differentiation but Does Not Improve Antigen-Specific Immune Responses. Clinical Cancer Research 2007, 13(16): 4840-4848.
- Albano D, Bosio G, Tomasini D, Bonù M, Giubbini R, Bertagna F: Metabolic behavior and prognostic role of pretreatment 18F-FDG PET/CT in gist. Asia Pac J Clin Oncol 2020, 16(5): e207-e215.
- Wang Z, Jiang Q, Dong C: Metabolic reprogramming in triple-negative breast cancer. Cancer Biol Med 2020, 17(1): 44-59.
- Aredo JV, Jamali A, Zhu J, Heater N, Wakelee HA, Vaklavas C, Anagnostou V, Lu J: Liquid Biopsy Approaches for Cancer Characterization, Residual Disease Detection, and Therapy Monitoring. Am Soc Clin Oncol Educ Book 2025, 45(3): e481114.
- Borger DR, Goyal L, Yau T, Poon RT, Ancukiewicz M, Deshpande V, Christiani DC, Liebman HM, Yang H, Kim H et al: Circulating oncometabolite 2-hydroxyglutarate is a potential surrogate biomarker in patients with isocitrate dehydrogenase-mutant intrahepatic cholangiocarcinoma. Clin Cancer Res 2014, 20(7): 1884-1890.
- Bobulescu IA, Pop LM, Mani C, Turner K, Rivera C, Khatoon S, Kairamkonda S, Hannan R, Palle K: Renal Lipid Metabolism Abnormalities in Obesity and Clear Cell Renal Cell Carcinoma. Metabolites 2021, 11(9): 608.
- Vovdenko S, Morozov A, Avraamova S, Alexandrov N, Zharkov N, Kozlov V, Kogan E, Bezrukov E: The role of glucose transporter type 1 (GLUT1) and carbonic anhydrase IX (CAIX) expression by prostate adenocarcinoma tissue in determining disease prognosis and effectiveness of radical treatment. Urology Herald 2022, 10(4): 13-20.
- Hansen AE, Gutte H, Holst P, Johannesen HH, Rahbek S, Clemmensen AE, Larsen MME, Schøier C, Ardenkjaer-Larsen J, Klausen TL et al: Combined hyperpolarized (13)C-pyruvate MRS and (18)F-FDG PET (hyperPET) estimates of glycolysis in canine cancer patients. Eur J Radiol 2018, 103: 6-12.
- Richter S, Gieldon L, Pang Y, Peitzsch M, Huynh T, Leton R, Viana B, Ercolino T, Mangelis A, Rapizzi E et al: Metabolome-guided genomics to identify pathogenic variants in isocitrate dehydrogenase, fumarate hydratase, and succinate dehydrogenase genes in pheochromocytoma and paraganglioma. Genet Med 2019, 21(3): 705-717.
- Trotter J, Pantel AR, Teo BK, Escorcia FE, Li T, Pryma DA, Taunk NK: Positron Emission Tomography (PET)/Computed Tomography (CT) Imaging in Radiation Therapy Treatment Planning: A Review of PET Imaging Tracers and Methods to Incorporate PET/CT. Adv Radiat Oncol 2023, 8(5): 101212.
- Mattoni S, Paccagnella A, Fanti S: The role of fluorine-18-fluorodeoxyglucose-positron emission tomography/computed tomography (18F-FDG-PET/CT) in staging and restaging of patients with uterine sarcomas: a systematic review. Gynecology and Pelvic Medicine 2021, 5: 7.
- Neumann K, Flavell R, Wilson DM: Exploring Metabolism In Vivo Using Endogenous (11)C Metabolic Tracers. Semin Nucl Med 2017, 47(5): 461-473.
- Wang ZJ, Ohliger MA, Larson PEZ, Gordon JW, Bok RA, Slater J, Villanueva-Meyer JE, Hess CP, Kurhanewicz J, Vigneron DB: Hyperpolarized (13)C MRI: State of the Art and Future Directions. Radiology 2019, 291(2): 273-284.
- Jhaveri KS, Elmi A, Hosseini-Nik H, Hedgire S, Evans A, Jewett M, Harisinghani M: Predictive Value of Chemical-Shift MRI in Distinguishing Clear Cell Renal Cell Carcinoma From Non-Clear Cell Renal Cell Carcinoma and Minimal-Fat Angiomyolipoma. AJR Am J Roentgenol 2015, 205(1): W79-86.
- Yu L, Chen X, Wang L, Chen S: The sweet trap in tumors: aerobic glycolysis and potential targets for therapy. Oncotarget 2016, 7(25): 38908-38926.
- Roviello G, De Gennaro I, Vascotto I, Venturi G, D'Angelo A, Winchler C, Guarino A, Cacioppo S, Modesti M, Mela MM et al: Hypoxia-Inducible Factor in Renal Cell Carcinoma: From Molecular Insights to Targeted Therapies. Genes (Basel) 2024, 16(1): 6.
- Wu H, Fu M, Wu M, Cao Z, Zhang Q, Liu Z: Emerging mechanisms and promising approaches in pancreatic cancer metabolism. Cell Death & Disease 2024, 15(8): 553.
- Yi M, Jiao D, Qin S, Chu Q, Wu K, Li A: Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol Cancer 2019, 18(1): 60.
- Qian Y, Yin Y, Zheng X, Liu Z, Wang X: Metabolic regulation of tumor-associated macrophage heterogeneity: insights into the tumor microenvironment and immunotherapeutic opportunities. Biomark Res 2024, 12(1): 1.
- Kurhanewicz J, Vigneron DB, Ardenkjaer-Larsen JH, Bankson JA, Brindle K, Cunningham CH, Gallagher FA, Keshari KR, Kjaer A, Laustsen C et al: Hyperpolarized (13)C MRI: Path to Clinical Translation in Oncology. Neoplasia 2019, 21(1): 1-16.
- Di Cosimo S, Ferretti G, Papaldo P, Carlini P, Fabi A, Cognetti F: Lonidamine: efficacy and safety in clinical trials for the treatment of solid tumors. Drugs Today (Barc) 2003, 39(3): 157-174.
- Curry L, Soleimani M: Belzutifan: a novel therapeutic for the management of von Hippel-Lindau disease and beyond. Future Oncol 2024, 20(18): 1251-1266.
- Lee CH, Motzer R, Emamekhoo H, Matrana M, Percent I, Hsieh JJ, Hussain A, Vaishampayan U, Liu S, McCune S et al: Telaglenastat plus Everolimus in Advanced Renal Cell Carcinoma: A Randomized, Double-Blinded, Placebo-Controlled, Phase II ENTRATA Trial. Clin Cancer Res 2022, 28(15): 3248-3255.
- Jones SF, Infante JR: Molecular Pathways: Fatty Acid Synthase. Clin Cancer Res 2015, 21(24): 5434-5438.
- Cheng HJ, Chen NF, Chen WF, Wu ZS, Sun YY, Teng WN, Su FW, Sung CS, Wen ZH: Intrathecal lactate dehydrogenase A inhibitors FX11 and oxamate alleviate chronic constriction injury-induced nociceptive sensitization through neuroinflammation and angiogenesis. J Headache Pain 2024, 25(1): 207.
- Hasanov E, Jonasch E: MK-6482 as a potential treatment for von Hippel-Lindau disease-associated clear cell renal cell carcinoma. Expert Opin Investig Drugs 2021, 30(5): 495-504.
- Mondal S, Roy D, Sarkar Bhattacharya S, Jin L, Jung D, Zhang S, Kalogera E, Staub J, Wang Y, Xuyang W et al: Therapeutic targeting of PFKFB3 with a novel glycolytic inhibitor PFK158 promotes lipophagy and chemosensitivity in gynecologic cancers. Int J Cancer 2019, 144(1): 178-189.
- Basheeruddin M, Qausain S: Hypoxia-Inducible Factor 1-Alpha (HIF-1α) and Cancer: Mechanisms of Tumor Hypoxia and Therapeutic Targeting. Cureus 2024, 16(10): e70700.
- Wray R, Mauguen A, Michaud L, Leithner D, Yeh R, Riaz N, Mirtcheva R, Sherman E, Wong R, Humm J et al: Development of (18)F-Fluoromisonidazole Hypoxia PET/CT Diagnostic Interpretation Criteria and Validation of Interreader Reliability, Reproducibility, and Performance. J Nucl Med 2024, 65(10): 1526-1532.
- Tan SK, Hougen HY, Merchan JR, Gonzalgo ML, Welford SM: Fatty acid metabolism reprogramming in ccRCC: mechanisms and potential targets. Nat Rev Urol 2023, 20(1): 48-60.
- Zhao Q, Lin X, Wang G: Targeting SREBP-1-Mediated Lipogenesis as Potential Strategies for Cancer. Front Oncol 2022, 12: 952371.
- Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, Kondo J, Coffey RJ, Johnson MO, Rathmell JC et al: Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med 2018, 24(2): 194-202.
- Kao TW, Bai GH, Wang TL, Shih IM, Chuang CM, Lo CL, Tsai MC, Chiu LY, Lin CC, Shen YA: Novel cancer treatment paradigm targeting hypoxia-induced factor in conjunction with current therapies to overcome resistance. J Exp Clin Cancer Res 2023, 42(1): 171.
- Yang WH, Qiu Y, Stamatatos O, Janowitz T, Lukey MJ: Enhancing the Efficacy of Glutamine Metabolism Inhibitors in Cancer Therapy. Trends Cancer 2021, 7(8): 790-804.
- Rai G, Brimacombe KR, Mott BT, Urban DJ, Hu X, Yang SM, Lee TD, Cheff DM, Kouznetsova J, Benavides GA et al: Discovery and Optimization of Potent, Cell-Active Pyrazole-Based Inhibitors of Lactate Dehydrogenase (LDH). J Med Chem 2017, 60(22): 9184-9204.
- Dall'Olio FG, Zrafi W, Roelants V, Ambrosini V, Fourquet A, Mitea C, Passiglia F, Bauckneht M, Bonardel G, Conci N et al: Metabolic Tumor Volume Assessed by 18F FDG-PET CT Scan as a Predictive Biomarker for Immune Checkpoint Blockers in Advanced NSCLC and Its Biological Correlates. Clin Cancer Res 2025, 31(2): 352-364.
- Ahmed N, Escalona R, Leung D, Chan E, Kannourakis G: Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: Perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Seminars in Cancer Biology 2018, 53: 265-281.
- Hao ZN, Tan XP, Zhang Q, Li J, Xia R, Ma Z: Lactate and Lactylation: Dual Regulators of T-Cell-Mediated Tumor Immunity and Immunotherapy. Biomolecules 2024, 14(12): 1646.
- You Y, Lai X, Pan Y, Zheng H, Vera J, Liu S, Deng S, Zhang L: Artificial intelligence in cancer target identification and drug discovery. Signal Transduction and Targeted Therapy 2022, 7(1): 156.
- Wahi K, Freidman N, Wang Q, Devadason M, Quek LE, Pang A, Lloyd L, Larance M, Zanini F, Harvey K et al: Macropinocytosis mediates resistance to loss of glutamine transport in triple-negative breast cancer. Embo j 2024, 43(23): 5857-5882.
- Audano M, Pedretti S, Crestani M, Caruso D, De Fabiani E, Mitro N: Mitochondrial dysfunction increases fatty acid β-oxidation and translates into impaired neuroblast maturation. FEBS Lett 2019, 593(22): 3173-3189.
- Rankin EB, Giaccia AJ: The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ 2008, 15(4): 678-685.
- Zhang Y, Shi M, Li M, Qin S, Miao D, Bai Y: Dynamic single-cell metabolomics reveals cell-cell interaction between tumor cells and macrophages. Nat Commun 2025, 16(1): 4582.
- Li AM, Ye J: Reprogramming of serine, glycine and one-carbon metabolism in cancer. Biochim Biophys Acta Mol Basis Dis 2020, 1866(10): 165841.
Annals of urologic oncology
p-ISSN: 2617-7765, e-ISSN: 2617-7773
 Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.