
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
Involvement of Epidermal Growth Factor Receptor (EGFR) Signaling in Renal Cell Carcinoma Progression and Therapeutic Implication
Ghulam Raza1, Kareem Khan21Department of Pathology, King Edward Medical University/Mayo Hospital, Lahore, Pakistan.
2Medical College, Aga Khan University, Karachi, Pakistan.
Correspondence: Ghulam Raza (Department of Pathology, King Edward Medical University/Mayo Hospital, Lahore, Pakistan; Email: raza26pk@gmail.com).
Annals of Urologic Oncology 2025, 8(1): 50-58. https://doi.org/10.32948/auo.2025.03.18
Received: 06 Jan 2025 | Accepted: 18 Mar 2025 | Published online: 22 Mar 2025
Key words renal cell carcinoma, epidermal growth factor receptor, VHL-HIF pathway, PI3K/Akt/mTOR, VEGF, angiogenesis
The molecular mechanism of RCC is complex. To improve the therapeutic intervention and increase the sensitivity, targeting potential prognostic biomarkers are the robust strategy. Meta-analysis identified three key genes including EGFR, FLT1, and EDN1 in RCC [4]. Among those EGFR is the most crucial biomarker and novel therapeutic target for treating RCC. EGFR is widely expressed in almost all cancer types including RCC and linked to disease progression, metastasis, poor outcome, and therapeutic resistance. VHL plays important functions in regulating the action of hypoxia-inducible factor (HIF) and ubiquitination. HIF is closely associated with angiogenesis. The action of VHL is suppressing tumors and loss of VHL and accumulation of HIF is the early sign of RCC. EGFR contributes to VHL loss in RCC through multiple pathways. Elevation of HIF in RCC is due to the upregulation of various growth factors including IGF, VEGF, EGF, and PDGF that activates PI3/AKT. Activation of PI3/AKT can be caused by stimulation of EGFR signaling. EGFR itself can also directly activate PI3K [5]. Additionally, EGFR suppresses autophagy in RCC leading to acceleration of RCC progression. EGFR also destabilizes E-cadherin, an epithelial marker essential for cellular adhesion. By attenuating E-cadherin, EGFR triggers the EMT initiation in RCC.
Elevated expression of EGFR in RCC was reported ranging from 50%-90% [6, 7]. The intracellular RTK domain of EGFR is autophosphorylated leading to the stimulation of multiple pathways in RCC including RAS-RAF-MEK-ERK and PI3K-PTEN-AKT pathways resulting in tumorigenesis in RCC. EGFR EGFR-mediated pathways are dysregulated in the RCC. Thus, proper characterization of EGFR localization in RCC and neighboring normal kidney tissues is important for EGFR-dependent anticancer therapeutic targeting. EGFR expression was assessed by immunohistochemistry (IHC) in the kidney tissues of 63 patients [8]. It was found that EGFR expression in the cell membrane was remarkably higher in the RCC tumor tissues in comparison to normal tissues. Conversely, EGFR expression in the cytoplasm was remarkably higher in the normal kidney tissues compared to RCC tumor tissues [8].
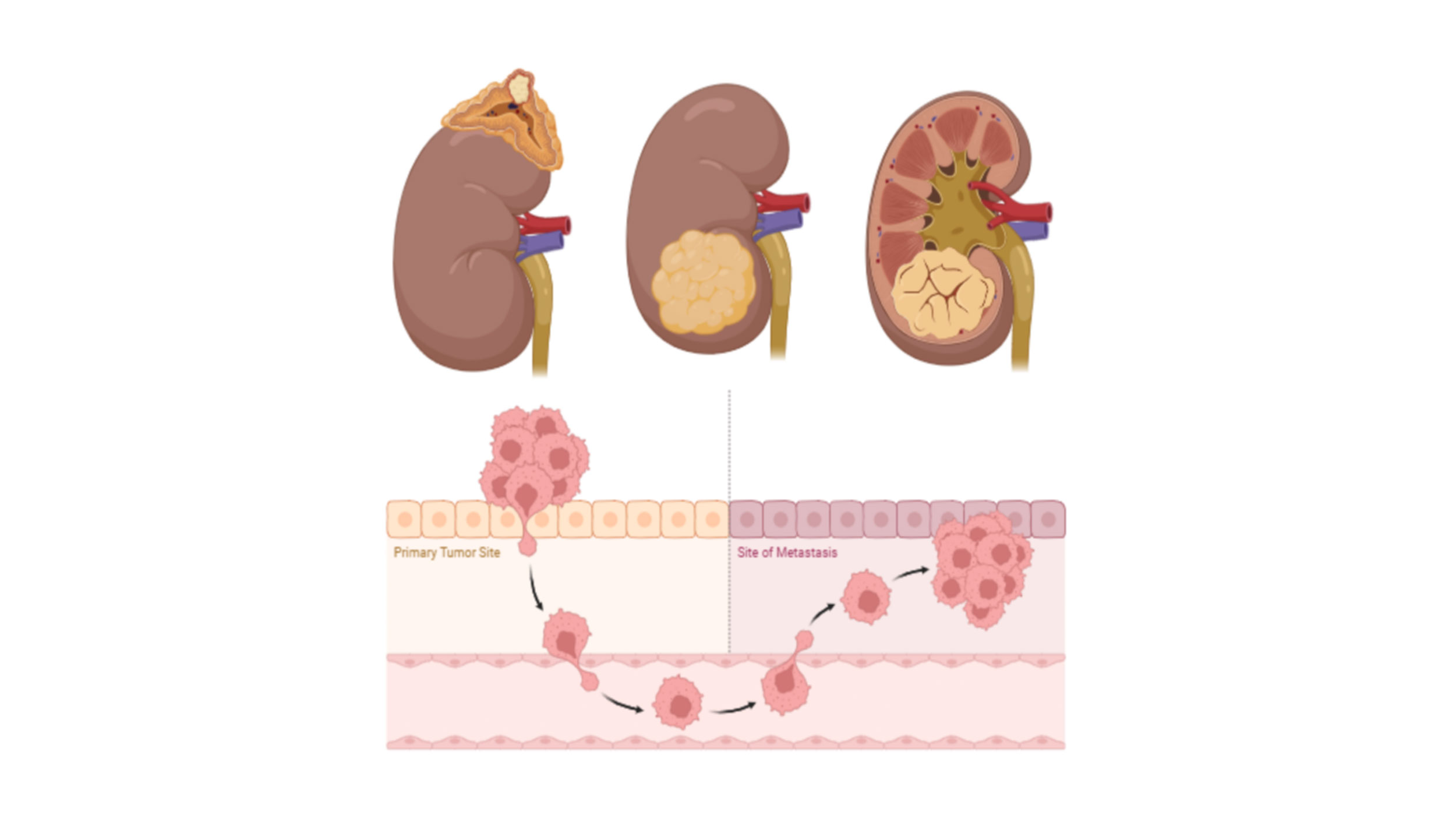 Figure 1. Formation of tumors in RCC. Overall survival rate is 5 years in RCC if the disease is localized in the kidney and the survival rate is dropped if the disease gets metastasized.
Figure 1. Formation of tumors in RCC. Overall survival rate is 5 years in RCC if the disease is localized in the kidney and the survival rate is dropped if the disease gets metastasized.
pVHL resides in the compartment of the cytoplasm and nucleus. pVHL consists of two mutational regions named alpha and beta domains. The α domain of pVHL directly binds to elongin C, and the beta domain directly binds to HIFα subunits [22] [23]. Different VHL mutations with respect to HIF regulation including Type 1, Type 2A, and Type 2B alleles. Among them, Type 2A pVHL mutant exhibited higher residual HIF-binding affinity compared to other mutants which is regarded as lower risk of RCC [24-26]. Patients detected with Type 1 and Type 2B VHL mutations form renal cysts having lost the WT allele of VHL and overproduction of numerous HIF gene products. Restoration of pVHL function by gene transfer does not alter cellular growth, although suppresses tumor formation in xenograft model [27, 28].
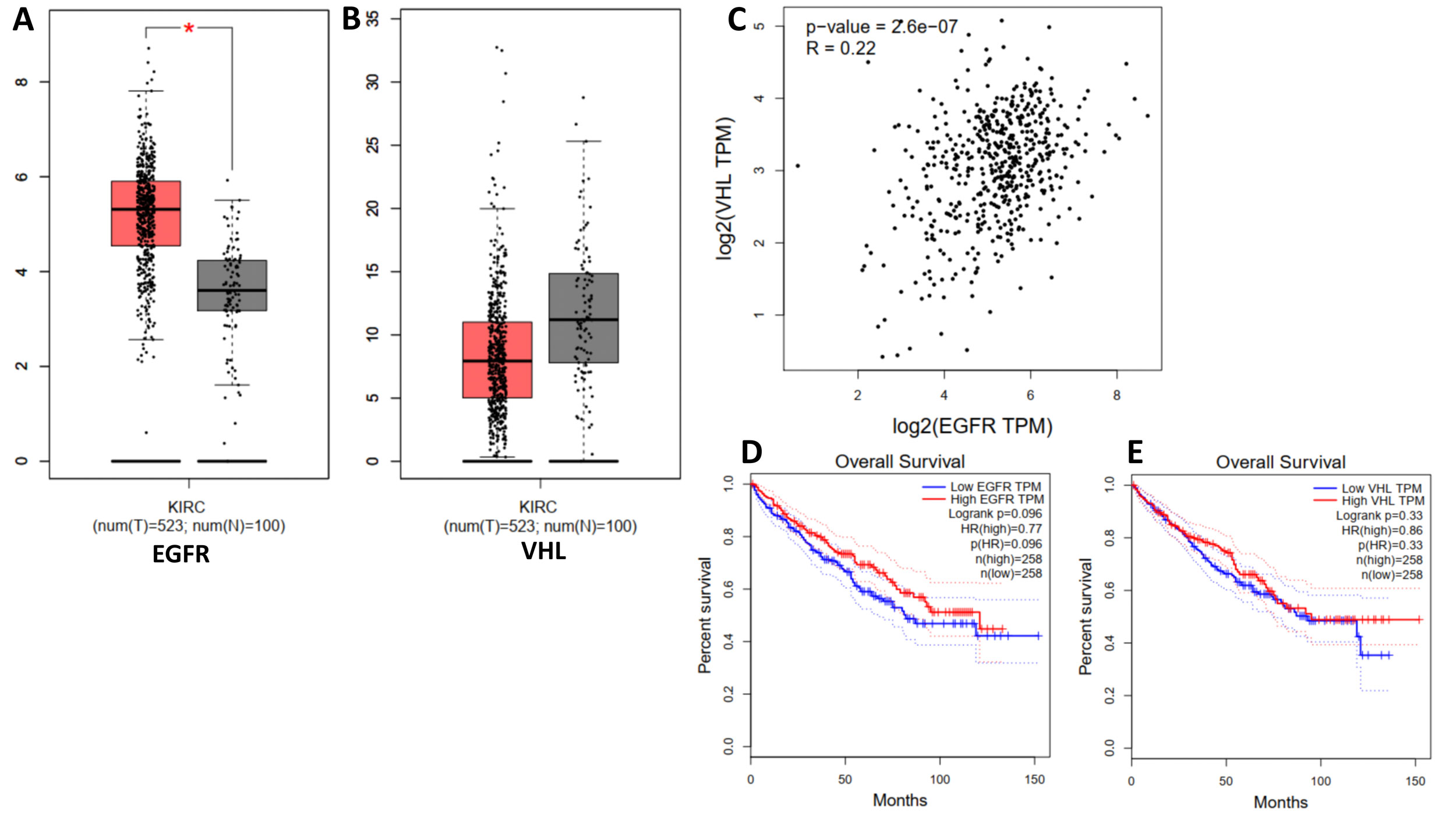 Figure 2. GEPIA plots. A: Box plot of EGFR expression in normal tissues (N) and tumor tissues of kidney renal clear cell carcinoma (T). B: Box plot of VHL expression in normal tissues (N) and tumor tissues of kidney renal clear cell carcinoma (T). C: Correlation of EGFR and VHL in RCC. D: The survival plot of low and high EGFR expression in RCC. E: Survival plot of low and high VHL expression in RCC.
Figure 2. GEPIA plots. A: Box plot of EGFR expression in normal tissues (N) and tumor tissues of kidney renal clear cell carcinoma (T). B: Box plot of VHL expression in normal tissues (N) and tumor tissues of kidney renal clear cell carcinoma (T). C: Correlation of EGFR and VHL in RCC. D: The survival plot of low and high EGFR expression in RCC. E: Survival plot of low and high VHL expression in RCC.
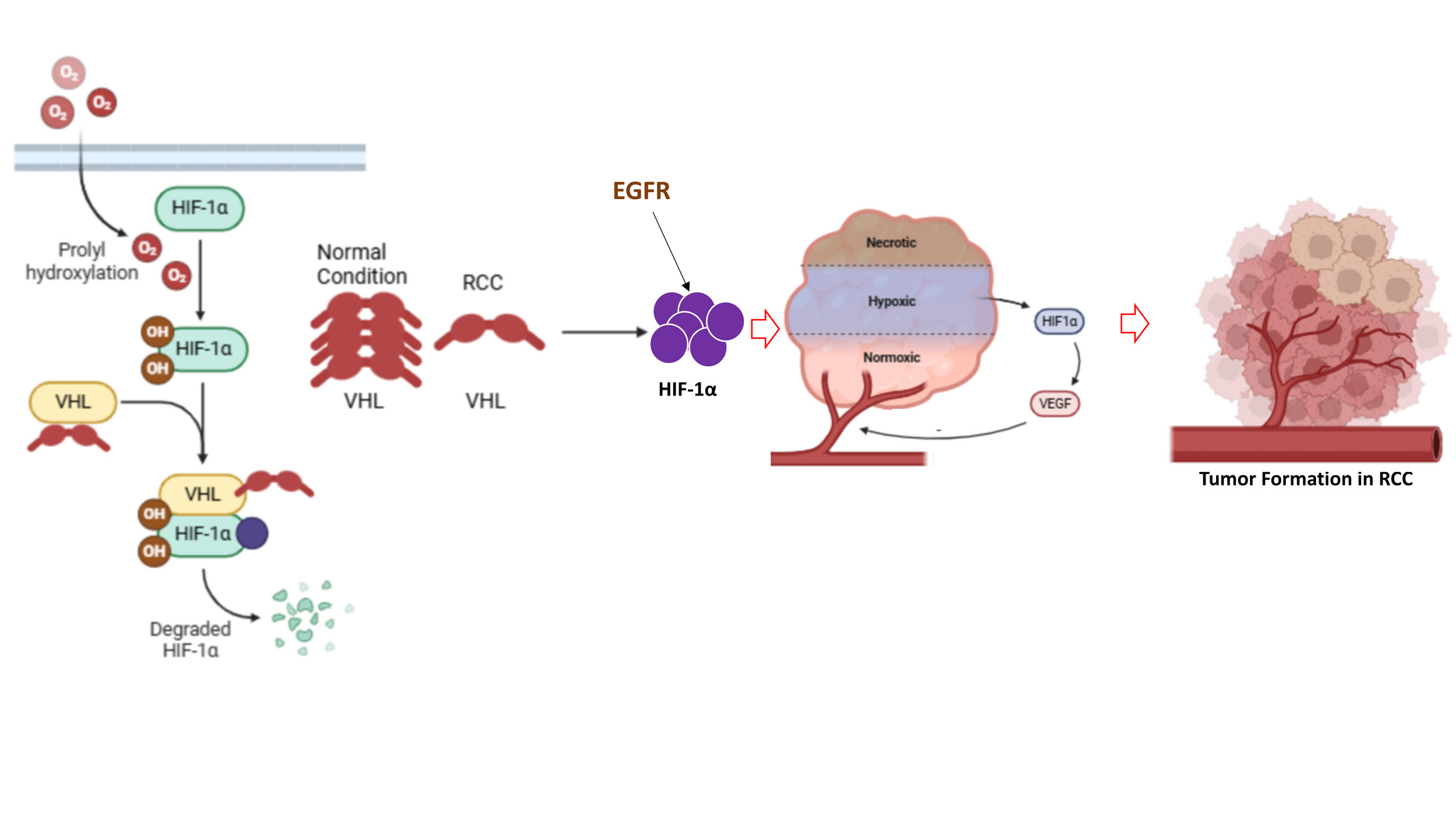 Figure 3. VHL-HIF1α pathway. Loss of VHL turns to elevation of HIF1α leads to initiation of RCC and tumor formation. EGFR contributes to the alteration of the VHL-HIF1α signaling pathway in RCC.
Figure 3. VHL-HIF1α pathway. Loss of VHL turns to elevation of HIF1α leads to initiation of RCC and tumor formation. EGFR contributes to the alteration of the VHL-HIF1α signaling pathway in RCC.
EGFR is a major contributor in the alteration of E-cadherin, claudins, and occludins through multiple pathways. Galectin-7 is a galectin family protein that plays a primary role in regulating cellular adhesion through directly interacting with E-cadherin [67-69]. E-cadherin is abundant at the adherent junctions of the epithelial cells. Several proteins including β-catenin maintain the integrity of the E-cadherin. EGFR also forms a complex with E-cadherin [70, 71]. However, it was reported that EGFR activation can suppress the function of E-cadherin [72, 73]. EGFR also decreases the stabilization of E-cadherin by enhancing E-cadherin internalization and altering the interaction of E-cadherin with the cytoskeleton by decreasing its expression on the cell surface [74]. It was revealed that upon EGF treatment in the cancer cells, β-catenin phosphorylation occurs at the Tyr654 and Tyr142 residues associated with reducing affinity to the E-cadherin leading to EMT progression [75, 76] in RCC and other cancer types. Sunitinib is an RTK inhibitor, widely used in treating cancer, particularly RCC. However, many patients remain unresponsive to the sunitinib treatment due to the major cause of EGFR-mediated EMT. In vitro study suggested that pEGFR was enhanced and followed by expression of mesenchymal markers was elevated upon treatment of sunitinib in the RCC cell line 786-O [77]. This study demonstrates that sunitinib mediated EMT via phosphorylation of EGFR in the 786-O cells which was alleviated by combination treatment with erlotinib. Besides, some other RCC cell lines such as Caki-1/SN were observed as resistant to sunitinib. Upon sunitinib treatment in the Caki-1/SN cells leading higher phosphorylation of EGFR [77].
Sunitinib is a selective RTK inhibitor including VEGFR and PDGFR. The potential antitumor action of Sunitinib was exerted by inhibiting angiogenesis. Sunitinib is successfully used in the RCC treatment and it is a primary intervention in RCC due to higher response rate. A phase I clinical trial of Sunitinib demonstrated notable antitumor actions in several patients with RCC.
Bevacizumab is used in the RCC treatment. The antitumor mechanism of bevacizumab is potent antiangiogenic action which neutralizes the vascular endothelial growth factor (VEGF). Human clear-cell renal carcinoma has a significantly higher blood vessel count compared to non-clear-cell renal carcinoma due to higher expression of VEGF. Bevacizumab mediated inhibition of angiogenesis leading to potent antitumor action in RCC [89].
Sorafenib is an oral multi-kinase inhibitor, and therapeutic agent for treating several cancers particularly RCC. Sorafenib possesses dual mechanisms including shrinkage of tumor growth and tumor anti-angiogenesis. In the clinical trial, sorafenib efficiently improved the total survival of the patients with lower toxicity. In the phase II clinical trial, sorafenib was well tolerated with less adverse effects. Sorafenib as a single therapeutic agent in RCC is very effective. However, to maximize the therapeutic efficacy sorafenib can be used with other therapeutic agents [90].
Cetuximab, a selective inhibitor of EGFR. The preclinical studies showed cetuximab has anti-tumor actions in RCC cell lines and remarkable xenograft tumor shrinkage in nude mice [91, 92]. However, in the phase II clinical trial patients with RCC did not exhibit objective response. The cetuximab response rate against RCC patients was much lower [93, 94].
None.
Ethical policy
Non applicable.
Availability of data and materials
All data generated or analysed during this study are included in this publication.
Author contributions
Ghulam Raza and Kareem Khan contributed to design of the work, data collection, and drafting the article. Ghulam Raza did the critical revision and approved the submission of the article.
Competing interests
The authors declare no competing interests.
Funding
None.
- Low G, Huang G, Fu W, Moloo Z, Girgis S: Review of renal cell carcinoma and its common subtypes in radiology. World J Radiol 2016, 8(5): 484-500.
- Sanchez DJ, Simon MC: Genetic and metabolic hallmarks of clear cell renal cell carcinoma. Biochim Biophys Acta Rev Cancer 2018, 1870(1): 23-31.
- Wettersten HI, Aboud OA, Lara PN, Jr., Weiss RH: Metabolic reprogramming in clear cell renal cell carcinoma. Nat Rev Nephrol 2017, 13(7): 410-419.
- Wang S, Yu ZH, Chai KQ: Identification of EGFR as a Novel Key Gene in Clear Cell Renal Cell Carcinoma (ccRCC) through Bioinformatics Analysis and Meta-Analysis. Biomed Res Int 2019, 2019: 6480865.
- Bussink J, van der Kogel AJ, Kaanders JHAM: Activation of the PI3-K/AKT pathway and implications for radioresistance mechanisms in head and neck cancer. Lancet Oncol 2008, 9(3): 288-296.
- Stumm G, Eberwein S, Rostock-Wolf S, Stein H, Pomer S, Schlegel J, Waldherr R: Concomitant overexpression of the EGFR and erbB-2 genes in renal cell carcinoma (RCC) is correlated with dedifferentiation and metastasis. Int J Cancer 1996, 69(1): 17-22.
- Moch H, Sauter G, Buchholz N, Gasser TC, Bubendorf L, Waldman FM, Mihatsch MJ: Epidermal growth factor receptor expression is associated with rapid tumor cell proliferation in renal cell carcinoma. Hum Pathol 1997, 28(11): 1255-1259.
- Pu Y-S, Huang C-Y, Kuo Y-Z, Kang W-Y, Liu G-Y, Huang AM, Yu H-J, Lai M-K, Huang S-P, Wu W-J et al: Characterization of membranous and cytoplasmic EGFR expression in human normal renal cortex and renal cell carcinoma. J Biomed Sci 2009, 16(1): 82.
- Cowey CL, Rathmell WK: VHL gene mutations in renal cell carcinoma: role as a biomarker of disease outcome and drug efficacy. Curr Oncol Rep 2009, 11(2): 94-101.
- Motzer RJ, Russo P: Systemic therapy for renal cell carcinoma. J Urol 2000, 163(2): 408-417.
- Bukowski RM: Cytokine therapy for metastatic renal cell carcinoma. Semin Urol Oncol 2001, 19(2): 148-154.
- Kaelin WG Jr: The von Hippel-Lindau tumor suppressor protein and clear cell renal carcinoma. Clin Cancer Res 2007, 13(2 Pt 2): 680s-684s.
- Dias F, Teixeira AL, Santos JI, Gomes M, Nogueira A, Assis J, Medeiros R: Renal cell carcinoma development and miRNAs: a possible link to the EGFR pathway. Pharmacogenomics 2013, 14(14): 1793-1803.
- Smaldone MC, Maranchie JK: Clinical implications of hypoxia inducible factor in renal cell carcinoma. Urol Oncol 2009, 27(3): 238-245.
- Hansen WJ, Ohh M, Moslehi J, Kondo K, Kaelin WG, Welch WJ: Diverse effects of mutations in exon II of the von Hippel-Lindau (VHL) tumor suppressor gene on the interaction of pVHL with the cytosolic chaperonin and pVHL-dependent ubiquitin ligase activity. Mol Cell Biol 2002, 22(6): 1947-1960.
- Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S, Negrier S, Szczylik C, Pili R, Bjarnason GA et al: Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 2009, 27(22): 3584-3590.
- Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA et al: Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007, 356(2): 125-134.
- He W, Batty-Stuart S, Lee JE, Ohh M: HIF-1α Hydroxyprolines Modulate Oxygen-Dependent Protein Stability Via Single VHL Interface With Comparable Effect on Ubiquitination Rate. J Mol Biol 2021, 433(22): 167244.
- Greijer AE, van der Wall E: The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J Clin Pathol 2004, 57(10): 1009-1014.
- Baldewijns MM, Thijssen VL, Van den Eynden GG, Van Laere SJ, Bluekens AM, Roskams T, van Poppel H, De Bruïne AP, Griffioen AW, Vermeulen PB: High-grade clear cell renal cell carcinoma has a higher angiogenic activity than low-grade renal cell carcinoma based on histomorphological quantification and qRT–PCR mRNA expression profile. Br J Cancer 2007, 96(12): 1888-1895.
- Ramakrishnan S, Anand V, Roy S: Vascular endothelial growth factor signaling in hypoxia and inflammation. J Neuroimmune Pharmacol 2014, 9(2): 142-160.
- Ivan M, Kaelin WG: The von Hippel–Lindau tumor suppressor protein. Curr Opin Genet Dev 2001, 11(1): 27-34.
- Ohh M, Park CW, Ivan M, Hoffman MA, Kim T-Y, Huang LE, Pavletich N, Chau V, Kaelin WG: Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel–Lindau protein. Nat Cell Biol 2000, 2(7): 423-427.
- Gossage L, Eisen T, Maher ER: VHL, the story of a tumour suppressor gene. Nat Rev Cancer 2015, 15(1): 55-64.
- Kaelin Jr WG: The von Hippel–Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer 2008, 8(11):865-873.
- Clifford SC, Cockman ME, Smallwood AC, Mole DR, Woodward ER, Maxwell PH, Ratcliffe PJ, Maher ER: Contrasting effects on HIF-1α regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum Mol Genet 2001, 10(10): 1029-1038.
- Azimi F, Naseripour M, Aghajani A, Kasraei H, Chaibakhsh S: The genetic differences between types 1 and 2 in von Hippel-Lindau syndrome: comprehensive meta-analysis. BMC Ophthalmol 2024, 24(1): 343.
- Hacker KE, Lee CM, Rathmell WK: VHL type 2B mutations retain VBC complex form and function. PLoS One 2008, 3(11): e3801.
- de Paulsen N, Brychzy A, Fournier MC, Klausner RD, Gnarra JR, Pause A, Lee S: Role of transforming growth factor-alpha in von Hippel--Lindau (VHL)(-/-) clear cell renal carcinoma cell proliferation: a possible mechanism coupling VHL tumor suppressor inactivation and tumorigenesis. Proc Natl Acad Sci U S A 2001, 98(4): 1387-1392.
- Szymańska K, Moore LE, Rothman N, Chow WH, Waldman F, Jaeger E, Waterboer T, Foretova L, Navratilova M, Janout V et al: TP53, EGFR, and KRAS mutations in relation to VHL inactivation and lifestyle risk factors in renal-cell carcinoma from central and eastern Europe. Cancer Lett 2010, 293(1): 92-98.
- Sakaeda T, Okamura N, Gotoh A, Shirakawa T, Terao S, Morioka M, Tokui K, Tanaka H, Nakamura T, Yagi M et al: EGFR mRNA is Upregulated, but Somatic Mutations of the Gene are Hardly Found in Renal Cell Carcinoma in Japanese Patients. Pharm Res 2005, 22(10): 1757-1761.
- Chan DA, Giaccia AJ: Hypoxia, gene expression, and metastasis. Cancer Metastasis Rev 2007, 26(2): 333-339.
- Smith K, Gunaratnam L, Morley M, Franovic A, Mekhail K, Lee S: Silencing of Epidermal Growth Factor Receptor Suppresses Hypoxia-Inducible Factor-2–Driven VHL−/− Renal Cancer. Cancer Res 2005, 65(12): 5221-5230.
- Paulsen ND, Brychzy A, Fournier MC, Klausner RD, Gnarra JR, Pause A, Pause A, Lee S: Role of transforming growth factor-alpha in von Hippel--Lindau (VHL)(-/-) clear cell renal carcinoma cell proliferation: a possible mechanism coupling VHL tumor suppressor inactivation and tumorigenesis. Proc Natl Acad Sci USA 2001, 98(4): 1387-1392.
- Stateva SR, Villalobo A: O-GlcNAcylation of the human epidermal growth factor receptor. Org Biomol Chem 2015, 13(30): 8196-8204.
- Tan DSW, Chong FT, Leong HS, Toh SY, Lau DP, Kwang XL, Zhang X, Sundaram GM, Tan GS, Chang MM et al: Long noncoding RNA EGFR-AS1 mediates epidermal growth factor receptor addiction and modulates treatment response in squamous cell carcinoma. Nat Med 2017, 23(10): 1167-1175.
- Qi HL, Li CS, Qian CW, Xiao YS, Yuan YF, Liu QY, Liu ZS: The long noncoding RNA, EGFR-AS1, a target of GHR, increases the expression of EGFR in hepatocellular carcinoma. Tumour Biol 2016, 37(1): 1079-1089.
- Wang A, Bao Y, Wu Z, Zhao T, Wang D, Shi J, Liu B, Sun S, Yang F, Wang L et al: Long noncoding RNA EGFR-AS1 promotes cell growth and metastasis via affecting HuR mediated mRNA stability of EGFR in renal cancer. Cell Death Dis 2019, 10(3): 154.
- Liu X-R, Zhou R-L, Zhang Q-Y, Zhang Y, Jin Y-Y, Lin M, Rui J-A, Ye D-X: Structure analysis and expressions of a novel tetratransmembrane protein, lysosoma-associated protein transmembrane 4 beta associated with hepatocellular carcinoma. World J Gastroenterol 2004, 10(11): 1555-1559.
- Tan X, Thapa N, Sun Y, Anderson RA: A kinase-independent role for EGF receptor in autophagy initiation. Cell 2015, 160(1-2): 145-160.
- Jutten B, Rouschop KM: EGFR signaling and autophagy dependence for growth, survival, and therapy resistance. Cell Cycle 2014, 13(1):42-51.
- Li X, Lu Y, Pan T, Fan Z: Roles of autophagy in cetuximab-mediated cancer therapy against EGFR. Autophagy 2010, 6(8): 1066-1077.
- Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW et al: EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013, 154(6): 1269-1284.
- Alers S, Löffler AS, Wesselborg S, Stork B: Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012, 32(1): 2-11.
- Kim J, Kundu M, Viollet B, Guan KL: AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011, 13(2): 132-141.
- Ewald JA, Wilkinson JC, Guyer CA, Staros JV: Ligand- and kinase activity-independent cell survival mediated by the epidermal growth factor receptor expressed in 32D cells. Exp Cell Res 2003, 282(2): 121-131.
- Weihua Z, Tsan R, Huang WC, Wu Q, Chiu CH, Fidler IJ, Hung MC: Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell 2008, 13(5): 385-393.
- Wang H, Wang Q, Wu Y, Lou J, Zhu S, Xu Y: Autophagy-related gene LAPTM4B promotes the progression of renal clear cell carcinoma and is associated with immunity. Front Pharmacol 2023, 14: 1118217.
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G: Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505(7484): 495-501.
- Jonasch E, Futreal PA, Davis IJ, Bailey ST, Kim WY, Brugarolas J, Giaccia AJ, Kurban G, Pause A, Frydman J et al: State of the science: an update on renal cell carcinoma. Mol Cancer Res 2012, 10(7): 859-880.
- Humtsoe JO, Kramer RH: Differential epidermal growth factor receptor signaling regulates anchorage-independent growth by modulation of the PI3K/AKT pathway. Oncogene 2010, 29(8): 1214-1226.
- Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, Cordon-Cardo C, Celeste Simon M, Rafii S, Pandolfi PP: PML inhibits HIF-1α translation and neoangiogenesis through repression of mTOR. Nature 2006, 442(7104): 779-785.
- Toschi A, Lee E, Gadir N, Ohh M, Foster DA: Differential dependence of hypoxia-inducible factors 1 alpha and 2 alpha on mTORC1 and mTORC2. J Biol Chem 2008, 283(50): 34495-34499.
- Akbani R, Ng PK, Werner HM, Shahmoradgoli M, Zhang F, Ju Z, Liu W, Yang JY, Yoshihara K, Li J et al: A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat Commun 2014, 5: 3887.
- Dawood M, Mills GB, Ding Z: Shrewd AKT regulation to survive. Oncoscience 2014, 1(2): 113-114.
- Gao M, Liang J, Lu Y, Guo H, German P, Bai S, Jonasch E, Yang X, Mills GB, Ding Z: Site-specific activation of AKT protects cells from death induced by glucose deprivation. Oncogene 2014, 33(6): 745-755.
- Cho D: Novel targeting of phosphatidylinositol 3-kinase and mammalian target of rapamycin in renal cell carcinoma. Cancer J 2013, 19(4): 311-315.
- Fruman DA, Rommel C: PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 2014, 13(2): 140-156.
- Liu H, Li X, Duan Y, Xie J-B, Piao X-L: Mechanism of gypenosides of Gynostemma pentaphyllum inducing apoptosis of renal cell carcinoma by PI3K/AKT/mTOR pathway. J Ethnopharmacol 2021, 271: 113907.
- Tang Z-L, Zhang K, Lv S-C, Xu G-W, Zhang J-F, Jia H-Y: LncRNA MEG3 suppresses PI3K/AKT/mTOR signalling pathway to enhance autophagy and inhibit inflammation in TNF-α-treated keratinocytes and psoriatic mice. Cytokine 2021, 148: 155657.
- Dorđević G, Matušan Ilijaš K, Hadžisejdić I, Maričić A, Grahovac B, Jonjić N: EGFR protein overexpression correlates with chromosome 7 polysomy and poor prognostic parameters in clear cell renal cell carcinoma. J Biomed Sci 2012, 19(1): 40.
- Sakaeda T, Okamura N, Gotoh A, Shirakawa T, Terao S, Morioka M, Tokui K, Tanaka H, Nakamura T, Yagi M et al: EGFR mRNA is upregulated, but somatic mutations of the gene are hardly found in renal cell carcinoma in Japanese patients. Pharm Res 2005, 22(10): 1757-1761.
- Chen K, Xiao H, Zeng J, Yu G, Zhou H, Huang C, Yao W, Xiao W, Hu J, Guan W et al: Alternative Splicing of EZH2 pre-mRNA by SF3B3 Contributes to the Tumorigenic Potential of Renal Cancer. Clin Cancer Res 2017, 23(13): 3428-3441.
- Zaman S, Hajiran A, Coba GA, Robinson T, Madanayake TW, Segarra DT, Chobrutskiy BI, Boyle TA, Zhou JM, Kim Y et al: Aberrant Epidermal Growth Factor Receptor RNA Splice Products Are Among the Most Frequent Somatic Alterations in Clear Cell Renal Cell Carcinoma and Are Associated with a Poor Response to Immunotherapy. Eur Urol Focus 2021, 7(2): 373-380.
- Li M, Cai L, Wang X, Yu Y, Jian W, Bao G, Gao Z, Guo J, Zhang J, Li C et al: RHBDD1 promotes proliferation, migration, invasion and EMT in renal cell carcinoma via the EGFR/AKT signaling pathway. Mol Med Rep 2021, 24(6): 826.
- Weygant N, Qu D, May R, Tierney RM, Berry WL, Zhao L, Agarwal S, Chandrakesan P, Chinthalapally HR, Murphy NT et al: DCLK1 is a broadly dysregulated target against epithelial-mesenchymal transition, focal adhesion, and stemness in clear cell renal carcinoma. Oncotarget 2015, 6(4): 2193-2205.
- Advedissian T, Proux-Gillardeaux V, Nkosi R, Peyret G, Nguyen T, Poirier F, Viguier M, Deshayes F: E-cadherin dynamics is regulated by galectin-7 at epithelial cell surface. Scientific Reports 2017, 7(1): 17086.
- Gendronneau G, Sidhu SS, Delacour D, Dang T, Calonne C, Houzelstein D, Magnaldo T, Poirier F: Galectin-7 in the control of epidermal homeostasis after injury. Mol Biol Cell 2008, 19(12): 5541-5549.
- Gendronneau G, Sanii S, Dang T, Deshayes F, Delacour D, Pichard E, Advedissian T, Sidhu SS, Viguier M, Magnaldo T et al: Overexpression of galectin-7 in mouse epidermis leads to loss of cell junctions and defective skin repair. PLoS One 2015, 10(3): e0119031.
- Hoschuetzky H, Aberle H, Kemler R: Beta-catenin mediates the interaction of the cadherin-catenin complex with epidermal growth factor receptor. J Cell Biol 1994, 127(5): 1375-1380.
- Pece S, Gutkind JS: Signaling from E-cadherins to the MAPK pathway by the recruitment and activation of epidermal growth factor receptors upon cell-cell contact formation. J Biol Chem 2000, 275(52): 41227-41233.
- Hazan RB, Norton L: The epidermal growth factor receptor modulates the interaction of E-cadherin with the actin cytoskeleton. J Biol Chem 1998, 273(15): 9078-9084.
- Fedor-Chaiken M, Hein PW, Stewart JC, Brackenbury R, Kinch MS: E-cadherin binding modulates EGF receptor activation. Cell Commun Adhes 2003, 10(2): 105-118.
- Ramírez Moreno M, Bulgakova NA: The Cross-Talk Between EGFR and E-Cadherin. Front Cell Dev Biol 2021, 9: 828673.
- Roura S, Miravet S, Piedra J, García de Herreros A, Duñach M: Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J Biol Chem 1999, 274(51): 36734-36740.
- Piedra J, Miravet S, Castaño J, Pálmer HG, Heisterkamp N, García de Herreros A, Duñach M: p120 Catenin-associated Fer and Fyn tyrosine kinases regulate beta-catenin Tyr-142 phosphorylation and beta-catenin-alpha-catenin Interaction. Mol Cell Biol 2003, 23(7): 2287-2297.
- Mizumoto A, Yamamoto K, Nakayama Y, Takara K, Nakagawa T, Hirano T, Hirai M: Induction of epithelial-mesenchymal transition via activation of epidermal growth factor receptor contributes to sunitinib resistance in human renal cell carcinoma cell lines. J Pharmacol Exp Ther 2015, 355(2): 152-158.
- Feng ZH, Fang Y, Zhao LY, Lu J, Wang YQ, Chen ZH, Huang Y, Wei JH, Liang YP, Cen JJ et al: RIN1 promotes renal cell carcinoma malignancy by activating EGFR signaling through Rab25. Cancer Sci 2017, 108(8): 1620-1627.
- Li Y, Jia Q, Zhang Q, Wan Y: Rab25 upregulation correlates with the proliferation, migration, and invasion of renal cell carcinoma. Biochem Biophys Res Commun 2015, 458(4): 745-750.
- Liu L, Ding G: Rab25 expression predicts poor prognosis in clear cell renal cell carcinoma. Exp Ther Med 2014, 8(4): 1055-1058.
- Ceresa BP: Regulation of EGFR endocytic trafficking by rab proteins. Histol Histopathol 2006, 21(9): 987-993.
- Ravaud A, de Clermont H, Pasticier G, Smith D, Vendrely V, Maire JP: Epithelial growth factor receptor (EGFR) pathway and renal cell carcinoma. Target Oncol 2007, 2(2): 99-105.
- Hutson TE, Quinn DI: Cytokine therapy: a standard of care for metastatic renal cell carcinoma? Clin Genitourin Cancer 2005, 4(3): 181-186.
- Minato N, Reid L, Cantor H, Lengyel P, Bloom BR: Mode of regulation of natural killer cell activity by interferon. J Exp Med 1980, 152(1): 124-137.
- Welsh RM, Yang H, Bukowski JF: The role of interferon in the regulation of virus infections by cytotoxic lymphocytes. Bioessays 1988, 8(1): 10-13.
- te Velde AA, de Waal Malefijt R, Huijbens RJ, de Vries JE, Figdor CG: IL-10 stimulates monocyte Fc gamma R surface expression and cytotoxic activity. Distinct regulation of antibody-dependent cellular cytotoxicity by IFN-gamma, IL-4, and IL-10. J Immunol 1992, 149(12): 4048-4052.
- Vuist WM, Visseren MJ, Otsen M, Bos K, Vyth-Dreese FA, Figdor CG, Melief CJ, Hekman A: Enhancement of the antibody-dependent cellular cytotoxicity of human peripheral blood lymphocytes with interleukin-2 and interferon alpha. Cancer Immunol Immunother 1993, 36(3): 163-170.
- Dorr RT: Interferon-alpha in malignant and viral diseases. A review. Drugs 1993, 45(2): 177-211.
- Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, Steinberg SM, Chen HX, Rosenberg SA: A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003, 349(5): 427-434.
- Adnane L, Trail PA, Taylor I, Wilhelm SM: Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol 2006, 407: 597-612.
- Perrotte P, Matsumoto T, Inoue K, Kuniyasu H, Eve BY, Hicklin DJ, Radinsky R, Dinney CP: Anti-epidermal growth factor receptor antibody C225 inhibits angiogenesis in human transitional cell carcinoma growing orthotopically in nude mice. Clin Cancer Res 1999, 5(2): 257-265.
- Prewett M, Rothman M, Waksal H, Feldman M, Bander NH, Hicklin DJ: Mouse-human chimeric anti-epidermal growth factor receptor antibody C225 inhibits the growth of human renal cell carcinoma xenografts in nude mice. Clin Cancer Res 1998, 4(12): 2957-2966.
- Yang XD, Jia XC, Corvalan JR, Wang P, Davis CG, Jakobovits A: Eradication of established tumors by a fully human monoclonal antibody to the epidermal growth factor receptor without concomitant chemotherapy. Cancer Res 1999, 59(6): 1236-1243.
- Yang XD, Jia XC, Corvalan JR, Wang P, Davis CG: Development of ABX-EGF, a fully human anti-EGF receptor monoclonal antibody, for cancer therapy. Crit Rev Oncol Hematol 2001, 38(1): 17-23.
Annals of urologic oncology
p-ISSN: 2617-7765, e-ISSN: 2617-7773
 Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.