
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
Synthetic Lethality in Prostate Cancer: Evaluating the Role of PARP Inhibitors in BRCA-Mutated mCRPC
Xinliang Xu1, Minna Liu21Department of Pain, Jining No.1 Peoples Hospital, Jining, 272011, China.
2Fundamental Medical Science Research Laboratories, The 940th Hospital Joint Logistics Support Forces of PLA, Lanzhou, 730030, China.
Correspondence: Minna Liu (Fundamental Medical Science Research Laboratories, The 940th Hospital Joint Logistics Support Forces of PLA, No. 333, South Binhe Middle Road, Lanzhou, Gan Su Province, P.R. China; Email: lmn2010@foxmail.com) and Xinliang Xu (Department of Pain, Jining No.1 Peoples Hospital, No. 6, Jiankang Road, Jining, Shan Dong Province, P.R. China; Email: 2466082197@qq.com).
Annals of Urologic Oncology 2025, 8(2): 88-96. https://doi.org/10.32948/auo.2025.05.11
Received: 11 May 2025 | Accepted: 23 Jun 2025 | Published online: 23 Jul 2025
Key words metastatic castration-resistant prostate cancer, androgen-receptor pathway inhibitors, synthetic lethality, immunotherapy
PARP inhibitors operationalize synthetic lethality by blocking catalytic PARylation and, critically, by “trapping” PARP on DNA, converting ordinarily reparable singlestrand lesions into lethal doublestrand breaks that BRCAdeficient cells cannot mend. Early proofofconcept came from the phase II TOPARPA/B trials, which demonstrated objective responses in BRCAmutated mCRPC [8], followed by the pivotal phase III PROfound study showing a radiographic progressionfree survival and overall survival advantage for olaparib with benefits most pronounced in the BRCA1/2 subset [9]. These data underpinned the 2020 food and drug administration (FDA) approvals of olaparib and rucaparib as mono-therapy for BRCAmutated mCRPC. The therapeutic paradigm has since shifted toward earlier, combinationbased approaches. In June 2023, the FDA approved talazoparib plus enzalutamide for firstline treatment of homologous recombination repairmutated mCRPC. Final overallsurvival results from the phase III TALAPRO2 trial, reported an 8.8 month median overall survival gain with talazoparib–enzalutamide versus placebo-enzalutamide in an unselected population, with the greatest absolute benefit again seen in BRCA1/2 carriers [2]. Despite these advances, several unmet needs persist: defining optimal sequencing with nextgeneration ARPIs now used earlier in the disease course, widening benefit beyond BRCA alterations, mitigating hematologic toxicity, and aDNA damage responseessing costeffectiveness in resourceconstrained settings.
Here, we critically evaluate the role of PARP inhibition within the specific context of BRCAmutated mCRPC. We first outline the genomic landscape of BRCA alterations in prostate cancer, then synthesize mechanistic and clinical data on PARP inhibitor mono-therapy and combinations, discuss predictive biomarkers, and examine emerging challenges such as resistance mechanisms, toxicity management and economic considerations. Finally, we highlight future directions aimed at integrating syntheticlethal strategies into precision oncology for advanced prostate cancer.
The principle of synthetic lethality, characterized as cell death caused by the concurrent loss of two distinct DNArepair mechanisms, has moved from conceptual genetics into routine oncology practice [6, 7]. In mCRPC, the most clinically exploitable syntheticlethal pair involves homologous recombination deficiency, most often driven by BRCA2 loss, and the singlestrand break repair machinery coordinated by PARP1/2 [23]. PARP1/2 detect singlestrand breaks through a zincfinger DNAbinding domain that recruits the catalytic domain to attach ADPribose polymers to histones and DNAbound proteins, thereby loosening chromatin and attracting the XRCC1ligase repair complex [24]. When PARP catalytic activity is blocked and the enzyme is “trapped” on DNA, unrepaired singlestrand breaks are converted into toxic doublestrand breaks (DSBs) during replication; BRCAdeficient cells, unable to perform accurate DSB repair, accumulate lethal genomic lesions, whereas BRCAproficient cells survive by restoring homologous recombination repair [2, 25]. Singlemolecule assays confirm that trapped PARP acts as a physical roadblock to the replication fork, producing stalled fork structures that collapse into DSBs unless BRCAmediated strand invasion repairs the break [26]. Four oral PARP inhibitors namely, olaparib, rucaparib, niraparib and talazoparib have been approved for mCRPC on the basis of this vulnerability, ushering in the first precision medicine option tailored to a defined molecular subgroup [27]. Biophysical analyses show that talazoparib stabilizes PARPDNA complexes up to 1000 fold more tightly than olaparib or rucaparib, while veliparib is a weak trapper despite potent catalytic inhibition [28]. Trapping potency correlates with both efficacy and hematologic toxicity. In comparative pharmacology work, talazoparib delivered the lowest 50 % inhibitory concentration (IC₅₀) across BRCA2knockout prostate cancer cell lines but also produced the highest rates of anemia and thrombocytopenia in clinical trials, whereas rucaparib and olaparib showed intermediate potency and toxicity, and niraparib fell in between [29, 30]. Beyond catalytic inhibition and trapping, PARP inhibitors also impair replicationfork stability, modulate transcriptioncoupled repair and activate cytosolic DNA sensing via cGAS–STING, all of which may synergise with immunotherapies explored in later sections [31]. Another important mechanistic nuance in prostate cancer is androgenreceptor (AR) crosstalk with DNA damage response pathways. AR signaling upregulates DNA damage response genes, including BRCA1/2; conversely, PARP1 functions as a transcriptional coactivator of AR. Preclinical models show that PARP blockade reduces AR chromatin occupancy, while AR antagonists increase reliance on PARPmediated repair, providing a biologic rationale for PARP–ARPI combinations now entering firstline therapy [25]. In the following subsections, we explore PARP inhibition-based mono- and combination therapies as synthetic lethality regimens in prostate cancer. Figure 1 shows that PARP inhibition-induced synthetic lethality in BRCA-mutated mCRPC.
PARP inhibitors as mono-therapy
Four oral PARP inhibitors namely, olaparib, rucaparib, niraparib and talazoparib have been approved for mCRPC [27]. The seminal phase II TOPARPA study and its histologyagnostic followup TOPARPB compared two olaparib doses in 98 biomarkerselected men with mCRPC; the higher 400 mg twicedaily regimen achieved a composite response in 54 % of patients with BRCA1/2 aberrations vs 4 % in homologous recombination repair wildtype controls [8, 32]. These results catalyzed the randomized phase III PROfound trial, in which olaparib (300 mg bid) outperformed physician’schoice ARPI (enzalutamide or abiraterone) in men whose tumors carried BRCA1/2 or ATM mutations. In the BRCA subgroup (n = 161), olaparib prolonged radiographic progressionfree survival to 9.8 months vs 3.0 months (HR 0.22) and extended overall survival to 20.1 months vs 14.4 months despite 66 % crossover [9]. This update confirmed an overall survival benefit across all ethnicities and priortherapy categories. On the other hand, the singlearm TRITON2 study first signaled activity of rucaparib 600 mg bid in postchemotherapy mCRPC, but objective responses in nonBRCA homologous recombination repair genes were negligible [33]. The phase III TRITON3 trial subsequently randomized men with chemotherapynaive mCRPC and BRCA1/2 or ATM alterations to rucaparib or docetaxel/ARPI. In the BRCA group (n = 405), rucaparib significantly delayed radiographic progression free survival (11.2 vs 6.4 months; HR 0.61) and improved time to symptomatic skeletal event; no radiographic progression free survival gain was seen in ATMmutant tumors, underscoring genespecific benefit. Grade ≥3 anemia (24 %) and fatigue (7 %) were common but manageable with dose modification [34]. Furthermore, the phase II GALAHAD study evaluated niraparib 300 mg daily in heavily pretreated men. Among 142 patients with biallelic BRCA1/2 defects, the objective response rate was 34 % with median radiographic progression free survival of 8.1 months and median overall survival of 13.0 months. In contrast, outcomes in nonBRCA DNA damage response genes were markedly poorer with objective response rate of 10 %, and radiographic progression free survival of 3.7 months [35]. Moreover, the highly potent trapper talazoparib demonstrated an objective response rate of 29 % and median radiographic progression free survival of 5.6 months in BRCAaltered tumors in the phase II TALAPRO1 study whereas nonBRCA responses were negligible [36]. A unifying theme across studies is the sharp dichotomy between BRCA1/2 and nonBRCA homologous recombination repair genes, supporting guideline recommendations that mono-therapy be restricted to BRCAmutated mCRPC. Secondly, outcomes correlate with biallelic loss as patients harbouring monoallelic germline variants without secondhit somatic events derive limited benefit from PARP inhibitors. Thirdly, haematologic toxicity rises with trapping potency (talazoparib > niraparib > olaparib ≈ rucaparib) and cumulative exposure, necessitating proactive full bloodcount monitoring and dose adjustments [12, 37]. Taken together, mono-therapy data firmly establish PARP inhibitors as a standard of care synthetic lethality inducing therapeutic for BRCAaltered, postARPI mCRPC, providing meaningful survival gains with manageable toxicity.
PARP inhibitors in combination therapies
Combination therapy has become the dominant clinical strategy for PARP inhibition in mCRPC. A growing body of laboratory work shows that AR signaling sustains DNA damage response gene expression, whereas PARP1 acts as an AR coactivator. Hence, dual blockade induces a deeper “BRCAness” and collapses replication forks even when homologous recombination repair is only partially impaired [38]. In the phase III PROpel trial, 399 firstline mCRPC patients received olaparib 300 mg bid with abiraterone/prednisone. The final pre-specified overall survival readout showed a nonsignificant numerical gain (median 42.1 vs 34.7 months; HR 0.81, p = 0.054) but confirmed durable radiographic progression-free survival benefit across all homologous recombination repair strata [39]. Posthoc analyses indicated the greatest absolute advantage in BRCAmutated tumors, whereas homologous recombination repair wildtype patients saw modest benefit at the cost of increased grade 3–4 anemia (16 %). Unlike PROpel, MAGNITUDE prospectively stratified 423 men by homologous recombination repair status before randomization. In the homologous recombination repair positive cohort, niraparib (200 mg daily) plus abiraterone prolonged radiographic progression free survival to 16.5 months vs 13.7 months with abiraterone alone (HR 0.73), significantly benefiting the BRCA1/2 subset [40]. The FDA granted this combination a breakthrough designation, and a fixeddose combination of niraparib plus abiraterone (Akeega®) is now licensed in Europe for homologous recombination repair mutated mCRPC [41]. The phase III TALAPRO2 study randomized 805 treatmentnaive patients to talazoparib 0.5 mg daily or placebo, each with enzalutamide. Overall survival analysis demonstrated a statistically significant 8.8month extension (45.8 vs 37.0 months; HR 0.80), with the largest relative effect in BRCA1/2 carriers (HR 0.54) [42]. Anemia and neutropenia were the key toxicities as 22 % discontinued talazoparib for adverse events. Talazoparibenzalutamide combination is now a firstline option for mCRPC irrespective of homologous recombination repair status. Overall, these finding suggest that combining PARP inhibitors with ARPIs is a potent synthetic lethality inducing strategy to limit the burden of BRCA-mutated mCRPC in clinics.
Immune checkpoint inhibitors alone have limited activity in mCRPC, however, these inhibitors can be combined with PARP inhibitors to boost the clinical benefits against prostate and other solid tumors [43, 44]. In KEYNOTE365 trial with combination treatment of olaparib and pembrolizumab, longterm followup of 79 postdocetaxel men revealed an objective response rate of 12 % and prostate-specific antigen declines in 48 %, with manageable immunerelated adverse events. Notably, these responses were enriched in DNA damage responsemutated tumors, but durability was modest (median radiographic progression free survival 5.4 months) [45]. A smaller investigatorinitiated trial of olaparib-durvalumab combination reported a 53 % composite response and median radiographic progression free survival of 16.1 months in 17 postARPI patients, with biomarker work suggesting correlation between response and low myeloidderived suppressorcell counts [46]. In a multicohort phase II CheckMate-9KD study, the rucaparib- nivolumab combination arm produced an overall response rate of 15 % and median radiographic progression free survival of 8.1 months in homologous recombination deficiencypositive mCRPC, again dominated by BRCA1/2 alterations. Grade ≥3 toxicity (mainly anemia, asthenia) occurred in 46 % [47]. Although, larger randomized trials (e.g., CASPAR) are under way to investigate PARP inhibition-induced synthetic lethality through combination therapies, predictive biomarkers are urgently needed to avoid unnecessary toxicity in nonresponders.
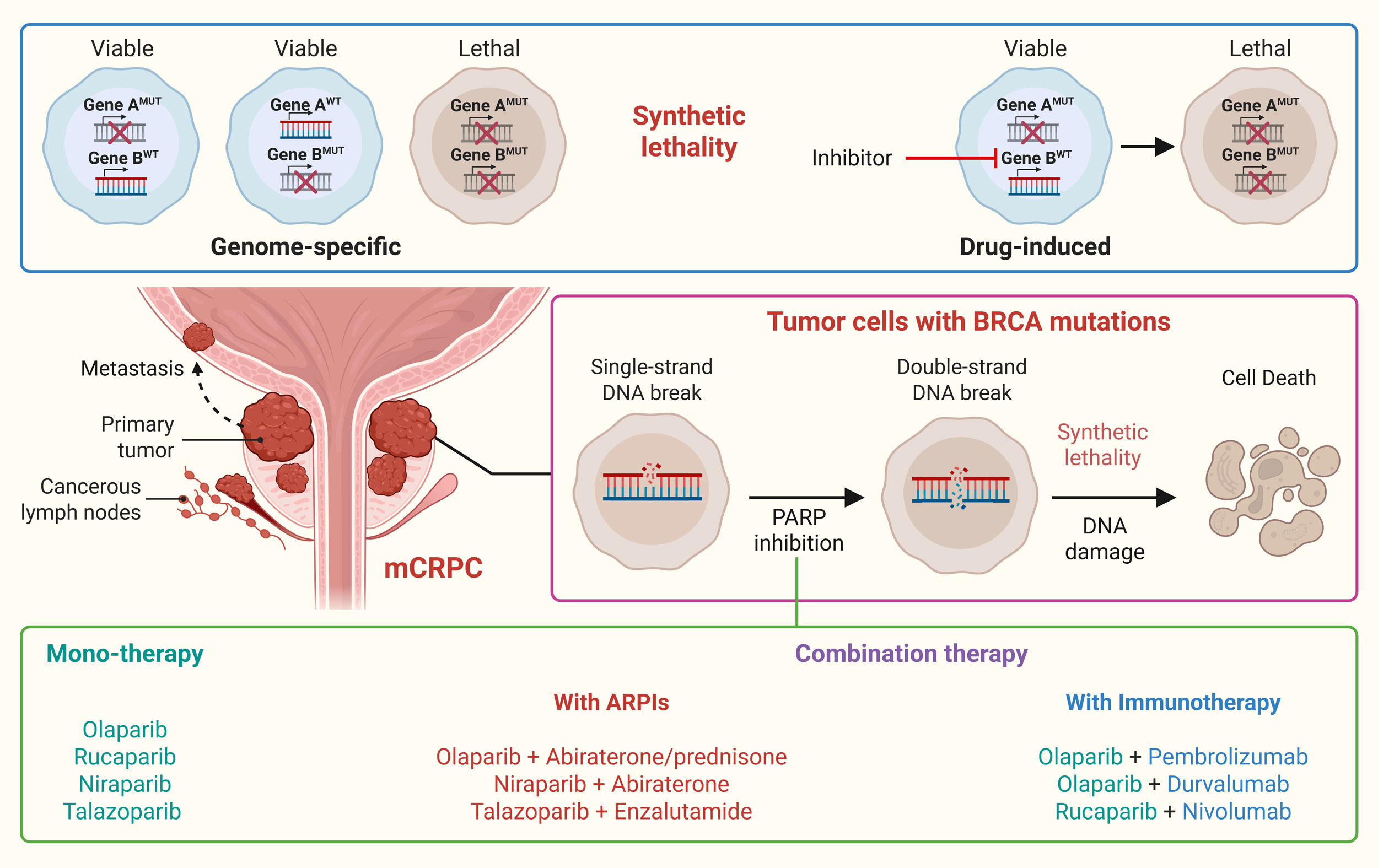 Figure 1. PARP inhibition-induced synthetic lethality in BRCA-mutated mCRPC. Synthetic lethality is characterized as cell death caused by the concurrent loss of two distinct targets/mechanisms which otherwise compensate for each other. Pharmacological inhibition of compensatory mechanism can also induce synthetic lethality. In this lines, targeting PARP in BRCA mutated mCRPC impairs single-strand repair, leading to double-strand breaks and resulting in synthetic lethality. Different PARP inhibitors have been approved and are being tested in clinical trials as monotherapy or in combination with ARPIs and immunotherapy as a treatment for mCRPC.
Figure 1. PARP inhibition-induced synthetic lethality in BRCA-mutated mCRPC. Synthetic lethality is characterized as cell death caused by the concurrent loss of two distinct targets/mechanisms which otherwise compensate for each other. Pharmacological inhibition of compensatory mechanism can also induce synthetic lethality. In this lines, targeting PARP in BRCA mutated mCRPC impairs single-strand repair, leading to double-strand breaks and resulting in synthetic lethality. Different PARP inhibitors have been approved and are being tested in clinical trials as monotherapy or in combination with ARPIs and immunotherapy as a treatment for mCRPC.
Therapy resistance against PARP inhibition
Resistance to PARP inhibitors can either be intrinsic (present before therapy) or acquired (during treatment) [56]. The bestdocumented mechanism is a reversion mutation that restores the open reading frame of a previously inactivating BRCA2 or BRCA1 allele, thereby reestablishing accurate doublestrandbreak repair. Serial ctDNA sequencing in subjects on olaparib or rucaparib showed that BRCA2 reversions arose in 39 % of progressors and were associated with a threefold hazard of death compared with patients who progressed without reversion [57]. Importantly, reversion events occur even in tumors with large genomic deletions via microhomologymediated endjoining and can involve multiple independent alleles within the same patient [58]. These findings explain why radiographic progression is often abrupt after an apparently durable response. Rewiring of replicationfork protection is another resistant mechanism. Pharmacologic inhibition of the ATR kinase, which is central to forkrestart signaling, can resensitize resistant clones, a concept now validated in BRCAmutant prostate models where ceralasertib plus olaparib restored cytotoxicity in vitro and in vivo [59]. In addition, point mutations or truncations in the PARP1 DNAbinding zinc finger reduce druginduced trapping while preserving catalytic function [60]. Because most commercial panels do not cover the relevant exons, PARP1 resistance mutations can be missed unless wholegenome sequencing or ctDNA deepamplicon panels are deployed. ABCfamily pumps, including ABCB1 (Pgp) and ABCC1, actively export PARP inhibitors such as olaparib and rucaparib, out of the cell [61]. Interference with nonhomologous endjoining (NHEJ) paradoxically restores homologous recombination repair in BRCA1 deficient cells. Prostate cancer organoids rendered PARP inhibitorresistant in vitro consistently lost SHLD2 or RIF1 expression, dismantling 53BP1shieldin–mediated endprotection and permitting resectionbased repair [62]. Chromatin modifiers, notably EZH2, modulate PARP inhibitor response. PARP inhibitor exposure itself induces a repressive heterochromatin landscape rich in H3K9me3, which dampens replication stress signaling and promotes survival. Pharmacologic EZH2 inhibitors reopen chromatin and re-sensitize resistant cultures, providing a rationale for earlyphase trials pairing talazoparib with tazemetostat [63, 64]. Prior PARP inhibitor exposure alters sensitivity to laterline treatments. In a multiinstitutional series of mCRPC patients receiving 177LuPSMA617, prior PARP inhibitor was associated with shorter progression-free survival, particularly among BRCA2 carriers, hinting at shared DNAdamage–response dependencies [65]. Further investigations are needed to fully understand the landscape of therapeutic resistance against PARP inhibitors in mCRPC to better device treatment strategies accordingly.
Toxicities and side-effects related to PARP inhibition in mCRPC
All four licensed PARP inhibitors share a broadly similar adverseevent spectrum driven by classspecific myelo-suppression and offtarget effects on rapidly proliferating tissues. For instance, grade ≥3 anemia occurred in 46 % of men receiving olaparib in the PROfound trial, making it the leading cause of dose interruption and the most common reason for transfusion [66]. Talazoparib, whose potent PARPtrapping activity translates into deeper marrow suppression, produced grade ≥3 anemia in 48 % and thrombocytopenia in 23 % of patients in the talazoparib–enzalutamide arm of TALAPRO2, with a 22 % permanent discontinuation rate [67]. Niraparib’s fixed daily dosing is associated with a higher incidence of hypertension [68]. Grade 2 cytopenia prompts oral iron supplementation if ferritin <30 µg/L and weekly counts; at grade 3, drug is held until recovery to ≤grade 1 and resumed at the next lower dose level. Granulocytecolonystimulatingfactor prophylaxis is not routinely recommended but can be deployed if neutropenia recurs despite two dose reductions [69]. Adding an ARPI deepens marrow suppression but introduces few new toxicities. In PROpel the olaparib–abiraterone arm showed a 16 % incidence of grade ≥3 anemia versus 4 % with abiraterone alone; hypertension and liverfunction abnormalities were unchanged [39]. Dermatologic toxicity, such as, photosensitivity, is infrequent and more pronounced with rucaparib, and sunprotection advice suffices in most cases [70]. PARP inhibitorassociated renal impairment typically manifests as a creatinine rise rather than a fall in glomerular filtration rate [71].
Cost ineffectiveness
The expansion of PARP inhibitors into firstline mCRPC raises unavoidable questions about affordability and equitable access. Cost in the United States hover around $200,000 for the median treatment duration seen of olaparib in PROpel study, whereas around $350,000 for the median treatment duration seen of Talazoparib in TALAPRO2 study [72]. Generic olaparib manufactured in India sells for approximately $3000 per month, still far above affordability thresholds in many low and middleincome countries. Outside highincome countries, access is further limited by delayed regulatory approvals and lack of reimbursement. Healthtechnologyassessment bodies in Latin America have approved olaparib only for ovariancancer indications, citing insufficient costeffectiveness data in prostate cancer [73].
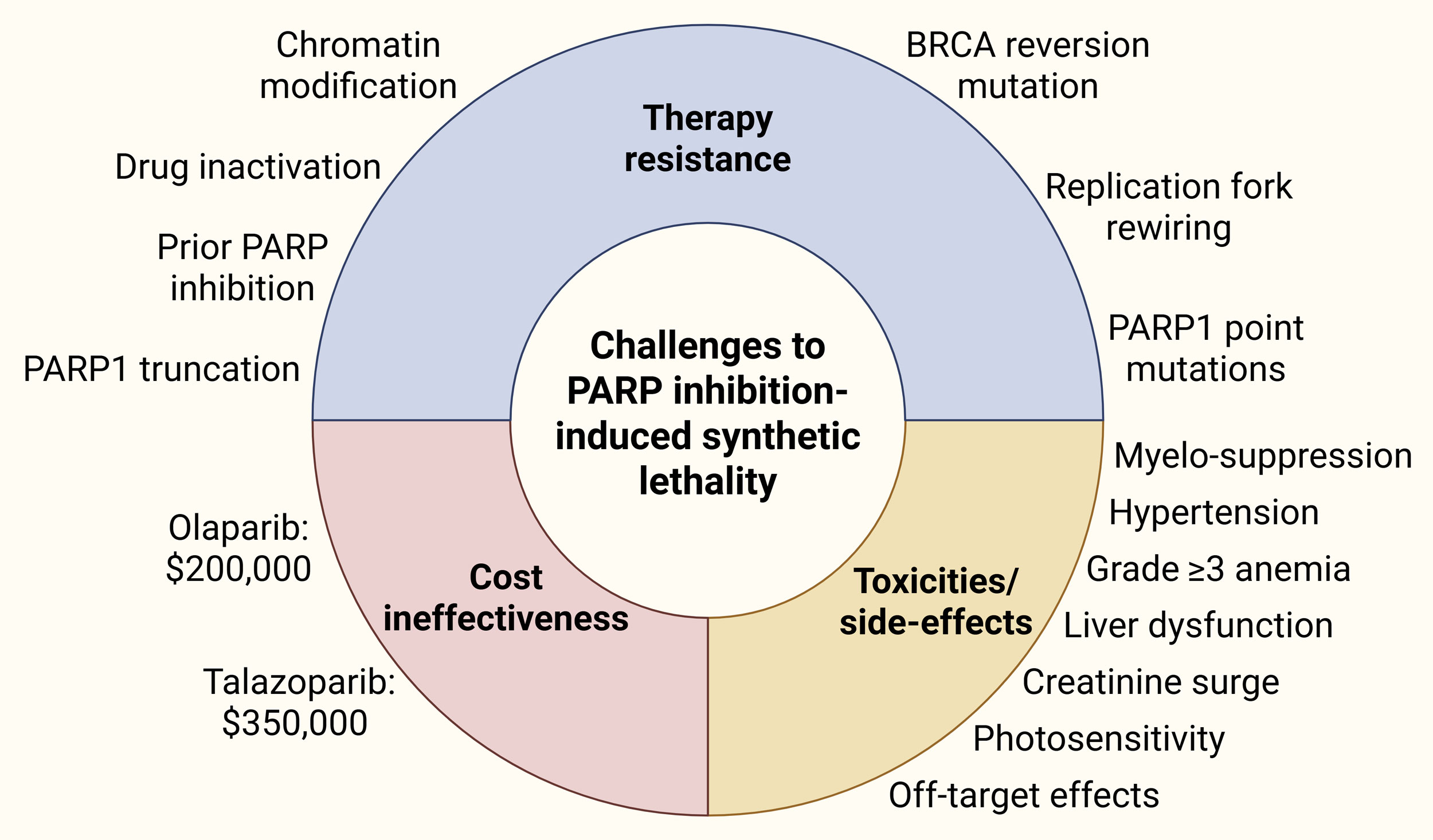 Figure 2. Challenges to PARP inhibition-induced synthetic lethality against mCRPC.
Figure 2. Challenges to PARP inhibition-induced synthetic lethality against mCRPC.
None.
Ethical policy
Non applicable.
Availability of data and materials
All data generated or analysed during this study are included in this publication.
Author contributions
Xinliang Xu contributed to design of the work, data collection, and drafting the article. Minna Liu did the critical revision and approved the submission of the article.
Competing interests
The authors declare no competing interests.
Funding
The study was supported by National Natural Science Foundation of China (82204746), Natural Science Foundation of Gansu Province (25JRRA810) and Natural Science Foundation of Lanzhou (2024-9-132).
- Rawla P: Epidemiology of Prostate Cancer. World J Oncol 2019, 10(2): 63-89.
- Bray F, Laversanne M, Sung H: Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2024, 74(3): 229-263.
- Kulasegaran T, Oliveira N: Metastatic Castration-Resistant Prostate Cancer: Advances in Treatment and Symptom Management. Curr Treat Options Oncol 2024, 25(7): 914-931.
- Sivakumar S, Lee JK, Moore JA, Hopkins J, Newberg JY, Madison R, Graf R, Schrock AB, Kobetz E, Vince R et al: Comprehensive genomic profiling and treatment patterns across ancestries in advanced prostate cancer: a large-scale retrospective analysis. Lancet Digit Health 2023, 5(6): e380-e389.
- Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, Garofalo A, Gulati R, Carreira S, Eeles R et al: Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N Engl J Med 2016, 375(5): 443-453.
- Helleday T: The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol 2011, 5(4): 387-393.
- Kaelin WG Jr.: The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 2005, 5(9): 689-698.
- Mateo J, Porta N, Bianchini D, McGovern U, Elliott T, Jones R, Syndikus I, Ralph C, Jain S, Varughese M et al: Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol 2020, 21(1): 162-174.
- Mateo J, de Bono JS: Olaparib for the Treatment of Patients With Metastatic Castration-Resistant Prostate Cancer and Alterations in BRCA1 and/or BRCA2 in the PROfound Trial. J Clin Oncol 2024, 42(5): 571-583.
- Khan HM, Young SMN, Russell EM, Esplin E, Korn WM, Cheng HH: Germline gene-specific associations in a large prostate cancer cohort. J Clin Oncol 2024, 42(16_suppl): 5102-5102.
- Khan HM, Cheng HH: Germline genetics of prostate cancer. Prostate 2022, 82 Suppl 1(Suppl 1): S3-s12.
- Olmos D, Lorente D, Alameda D, Cattrini C, Romero-Laorden N, Lozano R, Lopez-Casas PP, Jambrina A, Capone C, Vanden Broecke AM et al: Treatment patterns and outcomes in metastatic castration-resistant prostate cancer patients with and without somatic or germline alterations in homologous recombination repair genes. Ann Oncol 2024, 35(5): 458-472.
- Lukashchuk N, Barnicle A, Adelman CA, Armenia J, Kang J, Barrett JC, Harrington EA: Impact of DNA damage repair alterations on prostate cancer progression and metastasis. Front Oncol 2023, 13: 1162644.
- Wineland D, Le AN, Hausler R, Kelly G, Barrett E: Biallelic BRCA Loss and Homologous Recombination Deficiency in Nonbreast/Ovarian Tumors in Germline BRCA1/2 Carriers. JCO Precis Oncol 2023, 7: e2300036.
- Chakraborty G, Armenia J, Mazzu YZ, Nandakumar S, Stopsack KH: Significance of BRCA2 and RB1 Co-loss in Aggressive Prostate Cancer Progression. Clin Cancer Res 2020, 26(8): 2047-2064.
- Valsecchi AA, Dionisio R, Panepinto O, Paparo J, Palicelli A: Frequency of Germline and Somatic BRCA1 and BRCA2 Mutations in Prostate Cancer: An Updated Systematic Review and Meta-Analysis. Cancers (Basel) 2023, 15(9): 2435.
- Fettke H, Dai C, Kwan EM, Zheng T, Du P, Ng N, Bukczynska P, Docanto M, Kostos L, Foroughi S et al: BRCA-deficient metastatic prostate cancer has an adverse prognosis and distinct genomic phenotype. eBioMedicine 2023, 95: 104738.
- Stopsack KH, Vijai J: Germline DNA Damage Repair Variants and Prognosis of Patients with High-Risk or Metastatic Prostate Cancer. Clin Cancer Res 2025, 31(1): 122-129.
- Ma M, Zhu Y, Xiao C, Li R, Cao X, Kang R, Wang X, Li E: Novel insights into RB1 in prostate cancer lineage plasticity and drug resistance. Tumori 2024, 110(4): 252-263.
- Schaeffer EM, Srinivas S, Adra N, An Y, Bitting R, Chapin B, Cheng HH, D'Amico AV, Desai N, Dorff T et al: NCCN Guidelines® Insights: Prostate Cancer, Version 3.2024. J Natl Compr Canc Netw 2024, 22(3): 140-150.
- Shore N, Armstrong AJ, Barata P, Byrne L, Hafron J, Young S, Paller C, Wise DR, Ventii K, Samadi A et al: Implementing and Optimizing Universal Germline Genetic Testing for Patients With Prostate Cancer in Clinical Practice. Urology 2025, 199: 1-10.
- Cimadamore A, Cheng L: Circulating Tumor DNA Testing for Homology Recombination Repair Genes in Prostate Cancer: From the Lab to the Clinic. Int J Mol Sci 2021, 22(11): 5522.
- Fan Y, Liu Z, Chen Y, He Z: Homologous Recombination Repair Gene Mutations in Prostate Cancer: Prevalence and Clinical Value. Adv Ther 2024, 41(6): 2196-2216.
- Ali AAE, Timinszky G, Arribas-Bosacoma R, Kozlowski M, Hassa PO, Hassler M, Ladurner AG, Pearl LH, Oliver AW: The zinc-finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat Struct Mol Biol 2012, 19(7): 685-692.
- Longoria O, Beije N, de Bono JS: PARP inhibitors for prostate cancer. Semin Oncol 2024, 51(1-2): 25-35.
- Hopkins TA, Ainsworth WB, Ellis PA, Donawho CK, DiGiammarino EL, Panchal SC, Abraham VC, Algire MA, Shi Y, Olson AM et al: PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity in Both Cancer Cells and Healthy Bone Marrow. Mol Cancer Res 2019, 17(2): 409-419.
- Beatson EL, Chau CH, Price DK, Figg WD: PARP inhibitors on the move in prostate cancer: spotlight on Niraparib & update on PARP inhibitor combination trials. Am J Clin Exp Urol 2022, 10(4): 252-257.
- Rudolph J, Jung K, Luger K: Inhibitors of PARP: Number crunching and structure gazing. Proc Natl Acad Sci U S A 2022, 119(11): e2121979119.
- Zaman N, Kushwah AS, Badriprasad A, Chakraborty G: Unravelling the molecular basis of PARP inhibitor resistance in prostate cancer with homologous recombination repair deficiency. Int Rev Cell Mol Biol 2024, 389: 257-301.
- Bourlon MT, Valdez P, Castro E: Development of PARP inhibitors in advanced prostate cancer. Ther Adv Med Oncol 2024, 16: 17588359231221337.
- Rose M, Burgess JT, O'Byrne K, Richard DJ, Bolderson E: PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front Cell Dev Biol 2020, 8: 564601.
- Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N et al: DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med 2015, 373(18): 1697-1708.
- Abida W, Campbell D, Patnaik A, Bryce AH, Shapiro J, Bambury RM, Zhang J, Burke JM, Castellano D, Font A et al: Rucaparib for the Treatment of Metastatic Castration-resistant Prostate Cancer Associated with a DNA Damage Repair Gene Alteration: Final Results from the Phase 2 TRITON2 Study. Eur Urol 2023, 84(3): 321-330.
- Fizazi K, Piulats JM, Reaume MN, Ostler P, McDermott R, Gingerich JR, Pintus E, Sridhar SS, Bambury RM, Emmenegger U et al: Rucaparib or Physician's Choice in Metastatic Prostate Cancer. N Engl J Med 2023, 388(8): 719-732.
- Smith MR, Scher HI, Sandhu S, Efstathiou E, Lara PN, Jr., Yu EY, George DJ, Chi KN, Saad F, Ståhl O et al: Niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects (GALAHAD): a multicentre, open-label, phase 2 trial. Lancet Oncol 2022, 23(3): 362-373.
- de Bono JS, Mehra N, Scagliotti GV, Castro E, Dorff T, Stirling A, Stenzl A, Fleming MT, Higano CS, Saad F et al: Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): an open-label, phase 2 trial. The Lancet Oncology 2021, 22(9): 1250-1264.
- Shu Y, Ding Y, He X, Liu Y, Wu P, Zhang Q: Hematological toxicities in PARP inhibitors: A real-world study using FDA adverse event reporting system (FAERS) database. Cancer Med 2023, 12(3): 3365-3375.
- Kostos L, Tran B, Azad AA: Combination of PARP Inhibitors and Androgen Receptor Pathway Inhibitors in Metastatic Castration-Resistant Prostate Cancer. Drugs 2024, 84(9): 1093-1109.
- Saad F, Clarke NW, Oya M, Shore N, Procopio G, Guedes JD, Arslan C, Mehra N, Parnis F, Brown E et al: Olaparib plus abiraterone versus placebo plus abiraterone in metastatic castration-resistant prostate cancer (PROpel): final prespecified overall survival results of a randomised, double-blind, phase 3 trial. Lancet Oncol 2023, 24(10): 1094-1108.
- Rathkopf DE, Roubaud G, Chi KN, Efstathiou E, Attard G, Olmos D, Small EJ, Saad M, Castro E, Kim W et al: Patient-reported Outcomes for Patients with Metastatic Castration-resistant Prostate Cancer and BRCA1/2 Gene Alterations: Final Analysis from the Randomized Phase 3 MAGNITUDE Trial. Eur Urol 2024, https://doi.org/10.1016/j.eururo.2024.09.003. Epub ahead of print.
- Russu A, Hazra A, Tian H, Haddish-Berhane N, Perez Ruixo JJ: Population Pharmacokinetics of Niraparib/Abiraterone Acetate Administered as Single-Agent Combination and Dual-Acting Tablets Plus Prednisone for Metastatic Castration-Resistant Prostate Cancer. Adv Ther 2025, 42(4): 1860-1880.
- Agarwal N, Azad A, Carles J, Fay AP, Matsubara N, Szczylik C, Giorgi UD, Joung JY, Fong PCC, Voog E et al: Final overall survival (OS) with talazoparib (TALA) + enzalutamide (ENZA) as first-line treatment in unselected patients with metastatic castration-resistant prostate cancer (mCRPC) in the phase 3 TALAPRO-2 trial. J Clin Oncol 2025, 43(5_suppl): LBA18-LBA18.
- Zhou L, Wan Y, Zhang L, Meng H, Yuan L, Zhou S, Cheng W, Jiang Y: Beyond monotherapy: An era ushering in combinations of PARP inhibitors with immune checkpoint inhibitors for solid tumors. Biomed Pharmacother 2024, 175: 116733.
- Peyraud F, Italiano A: Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers (Basel) 2020, 12(6): 1502.
- Yu EY, Piulats JM, Gravis G, Fong PCC, Todenhöfer T, Laguerre B, Arranz JA, Oudard S, Massard C, Heinzelbecker J et al: Pembrolizumab plus Olaparib in Patients with Metastatic Castration-resistant Prostate Cancer: Long-term Results from the Phase 1b/2 KEYNOTE-365 Cohort A Study. Eur Urol 2023, 83(1): 15-26.
- Karzai F, VanderWeele D, Madan RA, Owens H, Cordes LM, Hankin A, Couvillon A, Nichols E, Bilusic M, Beshiri ML et al: Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J Immunother Cancer 2018, 6(1): 141.
- Fizazi K, Retz M, Petrylak DP, Goh JC, Perez-Gracia J, Lacombe L, Zschäbitz S, Burotto M, Mahammedi H, Gravis G et al: Nivolumab plus rucaparib for metastatic castration-resistant prostate cancer: results from the phase 2 CheckMate 9KD trial. J Immunother Cancer 2022, 10(8): e004761.
- Kazmi F, Shrestha N, Liu TFD, Foord T, Heesen P, Booth S, Dodwell D, Lord S, Yeoh KW, Blagden SP: Next-generation sequencing for guiding matched targeted therapies in people with relapsed or metastatic cancer. Cochrane Database Syst Rev 2025, 3(3): Cd014872.
- Fonseca NM, Maurice-Dror C, Herberts C: Prediction of plasma ctDNA fraction and prognostic implications of liquid biopsy in advanced prostate cancer. Nat Commun 2024, 15(1): 1828.
- Arce-Gallego S, Cresta Morgado P, Delgado-Serrano L, Simonetti S, Gonzalez M, Romero-Lozano P, Marmolejo D, Morales-Barrera R, Arnau GM, Semidey ME et al: Homologous recombination repair status in metastatic prostate cancer by next-generation sequencing and functional immunofluorescence. Cell Rep Med 2025, 6(2): 101937.
- Castroviejo-Bermejo M, Cruz C, Llop-Guevara A, Gutiérrez-Enríquez S, Ducy M, Ibrahim YH, Gris-Oliver A, Pellegrino B, Bruna A, Guzmán M et al: A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol Med 2018, 10(12): e9172.
- Goodall J, Mateo J, Yuan W, Mossop H, Porta N, Miranda S, Perez-Lopez R, Dolling D, Robinson DR, Sandhu S et al: Circulating Cell-Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discov 2017, 7(9): 1006-1017.
- Gutierrez AL, Halbwedl I, Sauer S, Regitnig P, Petru E, Seeböck R, Schubert S, Peternell C, Bodó K, Prein K et al: Robust Assessment of Homologous Recombination Deficiency Genomic Instability by OncoScan Microarrays. J Mol Diagn 2025, 27(6): 475-484.
- Saad F, Clarke NW, Oya M, Shore N, Procopio G, Guedes JD, Arslan C, Mehra N, Parnis F, Brown E et al: Olaparib plus abiraterone versus placebo plus abiraterone in metastatic castration-resistant prostate cancer (PROpel): final prespecified overall survival results of a randomised, double-blind, phase 3 trial. Lancet Oncol 2023, 24(10): 1094-1108.
- Richardson DL, Quintanilha JCF: Effectiveness of PARP Inhibitor Maintenance Therapy in Ovarian Cancer by BRCA1/2 and a Scar-Based HRD Signature in Real-World Practice. Clin Cancer Res 2024, 30(20): 4644-4653.
- Fojo T, Bates S: Mechanisms of resistance to PARP inhibitors--three and counting. Cancer Discov 2013, 3(1): 20-23.
- Loehr A, Hussain A, Patnaik A, Bryce AH, Castellano D, Font A, Shapiro J, Zhang J, Sautois B, Vogelzang NJ et al: Emergence of BRCA Reversion Mutations in Patients with Metastatic Castration-resistant Prostate Cancer After Treatment with Rucaparib. Eur Urol 2023, 83(3): 200-209.
- Pappas K, Ferrari M, Smith P, Nandakumar S, Khan Z, Young SB, LaClair J, Russo MV, Huang-Hobbs E, Schultz N et al: BRCA2 reversion mutation-independent resistance to PARP inhibition in prostate cancer through loss of function perturbations in the DNA pre-replication complex. bioRxiv 2025, https://doi.org/10.1101/2025.04.20.649668. Epub ahead of print.:2025.2004.2020.649668.
- Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, Todorova Kwan T, Morris R, Lauffer S, Nussenzweig A et al: ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev 2017, 31(3): 318-332.
- Zhu T, Zheng JY, Huang LL, Wang YH, Yao DF, Dai HB: Human PARP1 substrates and regulators of its catalytic activity: An updated overview. Front Pharmacol 2023, 14: 1137151.
- Lawlor D, Martin P, Busschots S, Thery J, O'Leary JJ, Hennessy BT, Stordal B: PARP Inhibitors as P-glyoprotein Substrates. J Pharm Sci 2014, 103(6): 1913-1920.
- Noordermeer SM, van Attikum H: PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol 2019, 29(10): 820-834.
- Choudhury AD, Xie W, Tewari A, Miyamoto DT, Kochupurakkal B, Ellis L, Bandel M, Leisner C, Shapiro G, D'Andrea AD et al: A phase Ia/Ib study of talazoparib in combination with tazemetostat in metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol 2022, 40(6_suppl): TPS195-TPS195.
- Zhang X, Huo X, Guo H, Xue L: Combined inhibition of PARP and EZH2 for cancer treatment: Current status, opportunities, and challenges. Front Pharmacol 2022, 13: 965244.
- Raychaudhuri R, Tuchayi AM, Low SK, Arafa AT, Graham LS, Gulati R, Pritchard CC, Montgomery RB, Haffner MC, Nelson PS et al: Association of Prior PARP Inhibitor Exposure with Clinical Outcomes after (177)Lu-PSMA-617 in Men with Castration-resistant Prostate Cancer and Mutations in DNA Homologous Recombination Repair Genes. Eur Urol Oncol 2025, https://doi.org/10.1016/j.euo.2025.01.002. Epub ahead of print.
- de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, Chi KN, Sartor O: Olaparib for Metastatic Castration-Resistant Prostate Cancer. N Engl J Med 2020, 382(22): 2091-2102.
- Azad AA, Fizazi K, Matsubara N, Saad F, De Giorgi U, Joung JY, Fong PCC, Jones RJ, Zschäbitz S, Oldenburg J et al: Talazoparib plus enzalutamide in metastatic castration-resistant prostate cancer: Safety analyses from the randomized, placebo-controlled, phase III TALAPRO-2 study. Eur J Cancer 2024, 213: 115078.
- Chen W, Xie J, Gao C, Zhang C, Fu Z, Shi C: Hypertension associated with niraparib in cancer patients: A pharmacovigilance analysis based on the FAERS database and meta-analysis of randomized controlled trials. Gynecol Oncol 2024, 182: 108-114.
- Tookman L, Krell J, Nkolobe B, Burley L, McNeish IA: Practical guidance for the management of side effects during rucaparib therapy in a multidisciplinary UK setting. Ther Adv Med Oncol 2020, 12: 1758835920921980.
- Mateos-Pujante A, Jiménez MC: Evaluation of phototoxicity induced by the anticancer drug rucaparib. Sci Rep 2022, 12(1): 3434.
- Bruin MAC, Korse CM, van Wijnen B, de Jong VMT, Linn SC, van Triest B, Rosing H, Beijnen JH, van den Broek D, Huitema ADR: A real or apparent decrease in glomerular filtration rate in patients using olaparib? Eur J Clin Pharmacol 2021, 77(2): 179-188.
- Benjamin DJ, Lester-Coll NH, Rezazadeh A: Financial toxicity from PARP inhibitors in castrate-resistant prostate cancer. J Clin Oncol 2024, 42(4_suppl): 54-54.
- Espinola N, Silvestrini C: Budget Impact Analysis of Olaparib for the Management of Patients with Homologous Recombination Repair (HRR)-Mutated Castration-Resistant Metastatic Prostate Cancer in Argentina. Pharmacoecon Open 2024, 8(5): 727-738.
- Herencia-Ropero A, Llop-Guevara A, Staniszewska AD, Domènech-Vivó J, García-Galea E, Moles-Fernández A, Pedretti F, Domènech H, Rodríguez O, Guzmán M et al: The PARP1 selective inhibitor saruparib (AZD5305) elicits potent and durable antitumor activity in patient-derived BRCA1/2-associated cancer models. Genome Med 2024, 16(1): 107.
- Chen L, Zou Y, Sun R, Huang M: Minimizing DNA trapping while maintaining activity inhibition via selective PARP1 degrader. Cell Death Dis 2024, 15(12): 898.
- Bazan Russo TD, Mujacic C, Di Giovanni E, Vitale MC, Ferrante Bannera C, Randazzo U, Contino S, Bono M: Polθ: emerging synthetic lethal partner in homologous recombination-deficient tumors. Cancer Gene Ther 2024, 31(11): 1619-1631.
- Zhou J, Gelot C, Pantelidou C: A first-in-class Polymerase Theta Inhibitor selectively targets Homologous-Recombination-Deficient Tumors. Nat Cancer 2021, 2(6): 598-610.
- Assaf ZJF, Zou W: A longitudinal circulating tumor DNA-based model associated with survival in metastatic non-small-cell lung cancer. Nat Med 2023, 29(4): 859-868.
- Shin SH, Cha S, Lee HY, Shin SH, Kim YJ, Park D, Han KY, Oh YJ, Park WY, Ahn MJ et al: Machine learning model for circulating tumor DNA detection in chronic obstructive pulmonary disease patients with lung cancer. Transl Lung Cancer Res 2024, 13(1): 112-125.
Annals of urologic oncology
p-ISSN: 2617-7765, e-ISSN: 2617-7773
 Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.