
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
Metabolic Supremacy Fuels Tumor Aggressiveness in Renal Cancer
Reham Gholam1, Muhammad Khalilzad21Department of Pharmacognosy, Daras Gah Mili Building, Islamic Azad University, ZIP area 11, Azarshahr Street, Karimkhan-e-Zand Avenue, Tehran 1477893780, Iran.
2Department of Basic Medical Science, Block 15, Islamic Azad University, ZIP area 11, Azarshahr Street, Karimkhan-e-Zand Avenue, Tehran 1477893780, Iran.
Correspondence: Muhammad Khalilzad (Department of Basic Medical Science, Block 15, Islamic Azad University, ZIP area 11, Azarshahr Street, Karimkhan-e-Zand Avenue, Tehran 1477893780, Iran; Email: Khalilzadah888@yahoo.com).
Annals of Urologic Oncology 2025, 8(1): 40-49. https://doi.org/10.32948/auo.2025.03.01
Received: 08 Jan 2025 | Accepted: 25 Feb 2025 | Published online: 02 Mar 2025
Key words renal cancer, hypoxia, Warburg effect, lipid metabolism, amino acid metabolism
First identified several decades ago, metabolic reprogramming has emerged as a primary focal point for new cancer treatments. It enables tumors to adapt biochemical pathways to satisfy escalated energy requirements and biosynthesis, thus presenting novel targets for intervention [10]. Early studies characterized the heightened glycolysis in cancer cells, known as the “Warburg effect”, as a reflection of inefficient energy generation [11]. However, modern evidence implicates oncogenic mutations in driving widespread metabolic alterations, influencing glucose uptake, lipid synthesis, and mitochondrial oxidative phosphorylation to advance tumorigenesis [12]. Even when oxygen is plentiful, cancer cells deplete local nutrient stores by favoring glycolysis over oxidative phosphorylation and establish an immunosuppressive environment that impairs T-cell function, thereby promoting tumor progression [13]. These metabolic abnormalities are particularly pronounced in ccRCC, with genetic disruptions triggering shifts in glucose metabolism, increased glutamine reliance, and mitochondrial dysfunction [14, 15]. The Warburg effect, exemplified by the bias toward rapid ATP generation while also producing building blocks vital for cellular proliferation, is a hallmark of ccRCC [16]. Hence, recognizing the relationship between metabolic dysregulation and ccRCC pathogenesis is essential for designing more effective treatments. Here, we explore the molecular mechanisms behind metabolic supremacy in ccRCC, focusing on dysfunctional hypoxic signaling, aberrant glucose metabolism, Warburg effect, dysregulated amino acid metabolism and elevated lipid metabolism as key culprits behind tumor aggressiveness. We also discuss the pharmacological interventions targeting these pathways, including agents that inhibit glycolysis, glutaminolysis, and fatty acid oxidation. By mapping the metabolic framework of ccRCC, this review aims to guide future research and therapeutic innovations intended to overcome tumor aggressive in renal cancer, critical to limit tumor burden and enhance patient survival in this malignancy.
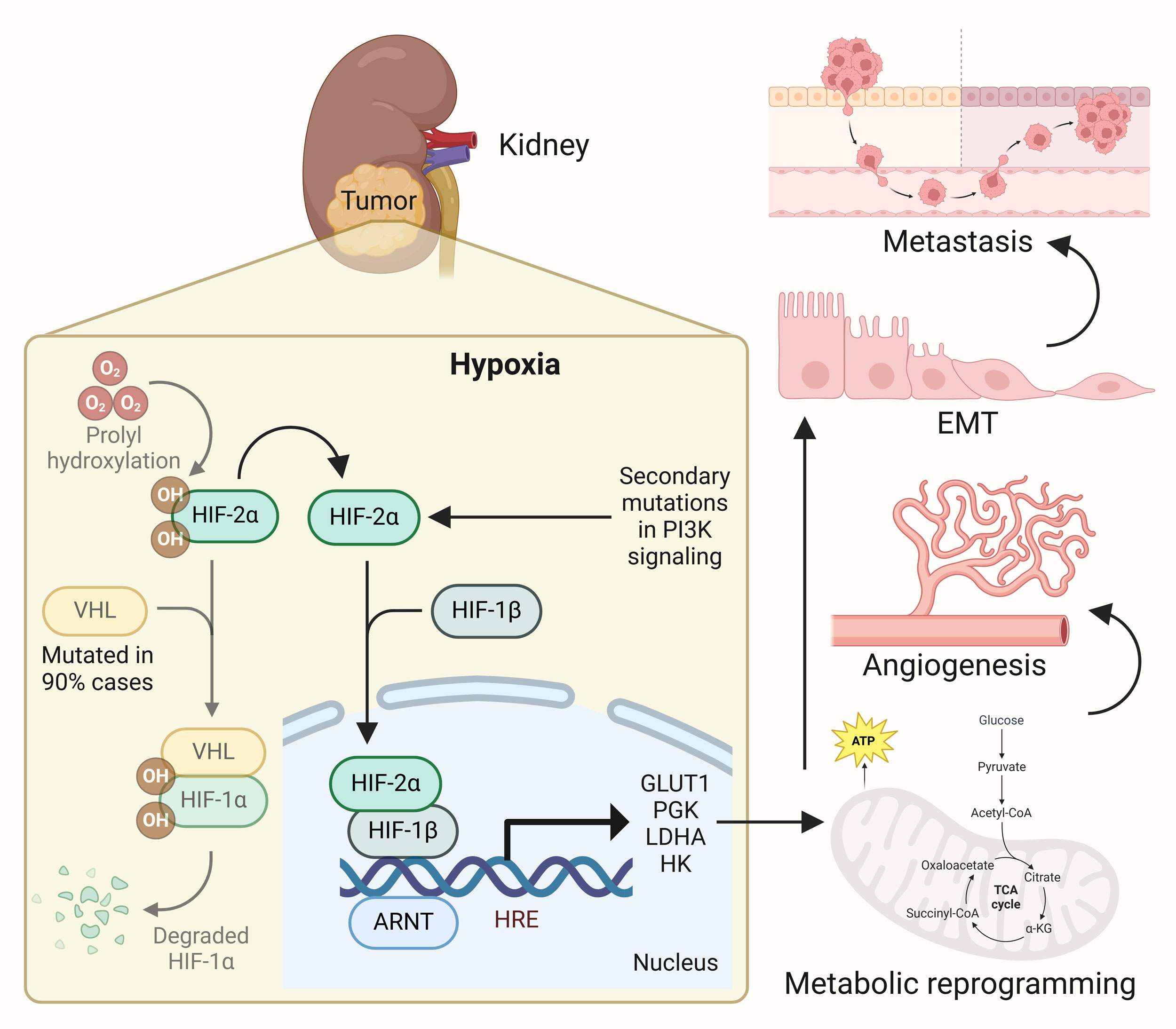 Figure 1. Hypoxia signaling induces metabolic supremacy in renal cancer. In response to poor oxygen supply in growing tumors in ccRCC, hypoxia signaling, primarily HIF-2α mediated one, gets activated leading to activation of plethora of genes involved in metabolic reprogramming. This results in onset of angiogenesis, tumor aggressiveness and progression, and metastatic spread of the disease.
Figure 1. Hypoxia signaling induces metabolic supremacy in renal cancer. In response to poor oxygen supply in growing tumors in ccRCC, hypoxia signaling, primarily HIF-2α mediated one, gets activated leading to activation of plethora of genes involved in metabolic reprogramming. This results in onset of angiogenesis, tumor aggressiveness and progression, and metastatic spread of the disease.
Glycolysis involves breaking glucose down into pyruvate [42]. Pyruvate enters the TCA cycle under aerobic conditions, supporting ATP generation along with the production of reduced nicotinamide adenine dinucleotide (NADH) and reduced flavin adenine dinucleotide (FADH2) [43]. Under anaerobic conditions, pyruvate is converted into lactate by fermentation, yielding ATP [44]. In ccRCC, another essential metabolic pathway is the pentose phosphate pathway, which generates glucose for lipid metabolism and nucleic acid synthesis by producing reduced nicotinamide adenine dinucleotide phosphate (NADPH) and ribose 5-phosphate [45]. The pentose phosphate pathway is significantly heightened in ccRCC, producing abundant NADPH necessary for preserving redox balance and safeguarding cancer cells from reactive oxygen species (ROS) damage [46]. This metabolic adjustment helps tumor cells mitigate oxidative stress and limit ROS-induced harm [47]. Additionally, the pentose phosphate pathway furnishes the five-carbon sugars required for nucleotide production, meeting the heightened demands of rapidly proliferating tumor cells [48]. Significant modifications also occur within the TCA cycle in ccRCC, contributing to its distinctive metabolic profile. Enzymes essential for refilling metabolic intermediates from other pathways are frequently reduced [49]. Of note, citrate and cis-aconitate are found in higher concentrations in ccRCC’s TCA cycle, whereas malate and fumarate are markedly decreased [15]. The drop in fumarate and malate is largely tied to the inhibition of succinate dehydrogenase (SDH), a mechanism that continuously diminishes fumarate and, by extension, malate [49]. This observation contradicts the typical notion of tumor tissues maintaining abundant fumarate. Overall, the abnormal regulation of glucose metabolism, primarily steered by HIF-1α and HIF-2α, plays a pivotal role in ccRCC aggressiveness. Collectively, overexpression of glycolytic enzymes, and alterations in the TCA cycle and pentose phosphate pathway grant ccRCC a metabolic advantage that propels its progression (Figure 2).
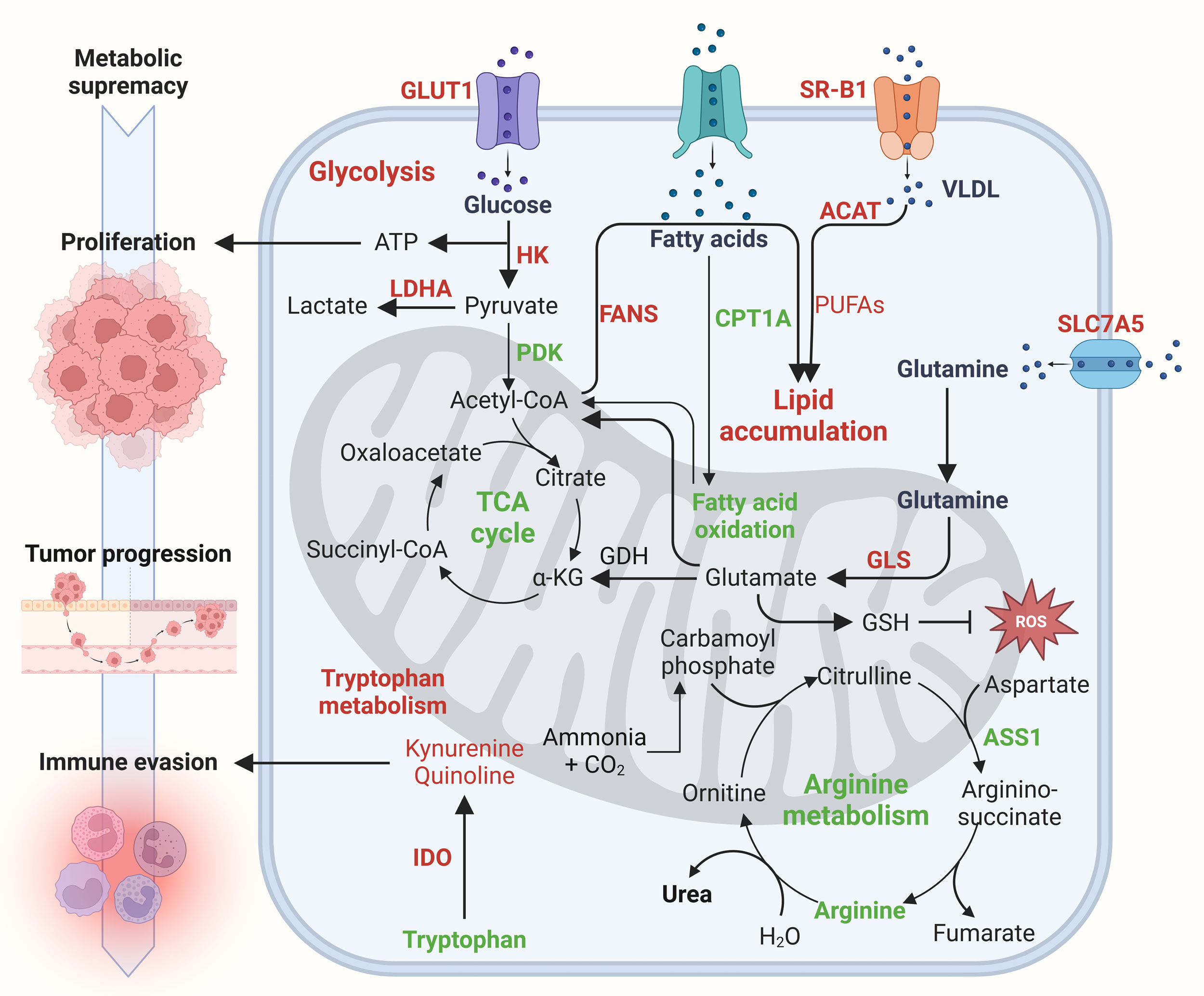 Figure 2. Metabolic supremacy drives tumor aggressiveness in renal cancer. Metabolic supremacy marked by elevated glucose, fatty acid, glutamine and tryptophan metabolism drives tumor aggressiveness in renal cancer. Glucose metabolism via glycolysis rather than oxidative phosphorylation (Warburg effect) fuels tumor proliferation and progression. Increased fatty acid and cholesterol uptake but reduced fatty acid oxidation leads to lipid accumulation which also serve as fuel for cancer progression. Meanwhile, enhanced glutamine metabolism counteracts oxidative stress via upregulating glutathione levels. Tumor cells limit arginine metabolism and rely on external arginine sources for rapid growth. Lastly, elevated tryptophan metabolism via IDO contribute to immune evasion.
Figure 2. Metabolic supremacy drives tumor aggressiveness in renal cancer. Metabolic supremacy marked by elevated glucose, fatty acid, glutamine and tryptophan metabolism drives tumor aggressiveness in renal cancer. Glucose metabolism via glycolysis rather than oxidative phosphorylation (Warburg effect) fuels tumor proliferation and progression. Increased fatty acid and cholesterol uptake but reduced fatty acid oxidation leads to lipid accumulation which also serve as fuel for cancer progression. Meanwhile, enhanced glutamine metabolism counteracts oxidative stress via upregulating glutathione levels. Tumor cells limit arginine metabolism and rely on external arginine sources for rapid growth. Lastly, elevated tryptophan metabolism via IDO contribute to immune evasion.
Tryptophan also critically modulates T cell-mediated immune responses to tumors. However, its excessive oxidation through the kynurenine pathway triggers T cell dysfunction and allows tumor cells to evade immune detection [66]. Metabolites in the kynurenine pathway actively suppress T cell activation, exacerbating immune escape. Within tumor-draining lymph nodes, elevated indoleamine 2,3-dioxygenase (IDO) activity creates an immunosuppressive milieu by prompting dendritic cells to inhibit T cells, thereby interfering with antigen recognition and immune function [67]. In ccRCC, immune checkpoint dysregulation is associated with increased IDO expression, which depletes tryptophan and activates the kynurenine pathway, ultimately supporting tumor survival by countering interferon-alpha (IFN-α) therapy and fostering immune suppression (Figure 2) [68, 69]. In addition, IDO overexpression has been closely tied to cancer metastasis: research in lung cancer cells reveals that higher IDO levels improve cell viability, whereas IDO inhibition diminishes it. In mouse models, administering lung cancer cells overexpressing IDO leads to more frequent metastases in the brain, liver, and bone [70]. These observations pinpoint IDO as a promising therapeutic target in ccRCC and other malignancies, warranting further exploration. Arginine metabolism is similarly disrupted in ccRCC, involving abnormalities in arginine transporters and metabolic enzymes, including arginase and arginine succinate synthase 1 (ASS1). Tumor cells often display reduced or missing ASS1 expression, an enzyme needed to convert citrulline into arginine, forcing cancer cells to rely on external arginine. Proteomic analyses of ccRCC biopsies support this dependency. Targeting arginine metabolism thus offers a potential therapeutic approach, as depriving tumor cells of this vital nutrient can suppress cancer progression. Research has shown that eliminating arginine selectively induces cell toxicity in ASS1-deficient tumors [71]. In summary, disrupted amino acid metabolism contribute to metabolic supremacy and tumor aggressiveness in renal cancer (Figure 2).
A particularly auspicious tactic targets the HIF-2α signaling pathway, a major downstream effector of the frequently mutated VHL tumor suppressor gene in ccRCC [90]. HIF-2α governs critical functions such as angiogenesis, cell proliferation, and metabolism, each substantially influencing tumor expansion and metastasis. Formerly deemed “undruggable” [91], HIF-2α has recently been shown to possess a structural vulnerability in its PAS-B domain. This discovery spurred the development of initial HIF-2α inhibitors like PT2399 and PT2385, which alter the PAS-B domain’s shape to block the formation of the HIF-2α/HIF-1β complex [92]. PT2399 has even surpassed sunitinib in certain models and remained effective against sunitinib-resistant tumors, although prolonged therapy can induce resistance via mutations in the binding site or HIF-1β [93], and it does not fully suppress all HIF-2α target genes. PT2385, meanwhile, showed a favorable safety profile and minimal toxicity in phase I trials, with a complete response observed in 2% of patients, partial responses in 12%, and stable disease in 52% [94]. In response to the shortcomings of these first-generation inhibitors, second-generation HIF-2α antagonists have been created, such as PT2977 (also known as MK-6482 or belzutifan) [95]. PT2977 targets a region adjacent to the PAS-B domain, triggering a conformational change that disrupts gene interactions. It also boasts low lipophilicity, high oral bioavailability, and an encouraging safety profile. In a phase II trial, a daily dose of 120 mg resulted in a 49% objective response rate among ccRCC patients, with primarily mild and manageable side effects [96, 97]. Consequently, newer HIF-2α inhibitors, particularly PT2977, have emerged as promising targeted therapeutics for ccRCC, offering improved potency and tolerability compared to older treatments.
Cancer cells also exploit glutamine metabolism to support energy generation, redox balance, and the synthesis of essential macromolecules. In ccRCC, GLS replenishes the TCA cycle and modestly drives cell proliferation [12]. The GLS inhibitor CB-839 has demonstrated strong anticancer activity in preclinical models. In animal studies, combining CB-839 with Everolimus, an mTOR inhibitor commonly used in ccRCC, enhanced antitumor efficacy. Clinical exploration of this combination, however, remains sparse, emphasizing the need for more extensive safety and effectiveness data [98, 99]. Furthermore, some cancers exhibit elevated arginine dependence due to deficient ASS1, thereby increasing their reliance on external arginine [100]. Since arginine is pivotal for nitric oxide production and protein biosynthesis, reducing circulating arginine through ADI-PEG20 (a PEG-conjugated arginine deaminase) has been proposed as a strategy to constrain tumor growth in ccRCC. That said, the re-expression of ASS1 may diminish its impact [12]. Clinical data point to good tolerance of ADI-PEG20 and suggest it may overcome drug resistance in cancers that rely heavily on arginine [101]. Additionally, encouraging results have been reported for ADI-PEG20 in non-small cell lung cancer, acute myeloid leukemia, and uveal melanoma [102, 103]. Further research will be crucial for refining combination regimens and determining how best to avert resistance. The enzyme IDO degrades tryptophan via the kynurenine pathway, contributing to an immunosuppressive tumor environment by lowering tryptophan levels, thereby impeding T-cell function and promoting metastasis [69]. IDO inhibition has thus become an appealing immunotherapeutic avenue. The selective IDO inhibitor Epacadostat was found in preclinical studies to enhance the response of tumor-specific T cells [104], though clinical outcomes have been mixed, showing toxicity issues and moderate efficacy at best. Early-phase trials combining Epacadostat with the PD-1 inhibitor pembrolizumab showed limited but notable antitumor responses in advanced solid tumors; however, more research is needed to fully establish its clinical benefit [105]. Meanwhile, Navoximod has displayed acceptable tolerability and moderate bioavailability at 800 mg twice daily. Although Navoximod monotherapy showed limited impact, pairing it with Atezolizumab produced encouraging safety profiles and measurable antitumor activity, with ongoing trials investigating its broader therapeutic potential [106-108]. Additional IDO inhibitors, such as KHK2455, LY3381916, and MK-7162, are in clinical studies to evaluate their safety and efficacy [67]. Moving forward, it will be essential to refine combination therapies and dosing strategies for IDO-targeted treatments. When considered alongside approaches like glycolysis inhibition, HIF-2α antagonism, and interventions involving lipid, glutamine, and arginine pathways, the growing range of metabolic therapies holds real promise for improving outcomes in ccRCC.
Genetic alterations in ccRCC also impact lipid metabolism, which is integral to tumor proliferation. FAS overexpression, a common finding, elevates intracellular fatty acid concentrations, fueling cancer growth and affecting post-translational modifications. Fatty acids are critical for both energy production and the maintenance of redox balance [109]. These insights have led researchers to propose inhibiting fatty acid synthesis as a therapeutic approach, supported by studies correlating greater FAS expression with higher tumor aggressiveness and worse clinical outcomes [110]. Preclinical evaluations indicate that the FAS inhibitor C75 can limit ccRCC cell proliferation and aggressiveness [111]. Another agent, TVB-2640, has shown promise in clinical contexts. A phase I investigation reported reduced fatty acid production in patients with non-small cell lung cancer, and follow-up trials in breast and ovarian cancers confirmed its efficacy and generally mild dermatological and ocular side effects [112]. TVB-2640 is currently undergoing evaluation in numerous cancer trials, including studies focused on ccRCC, suggesting that FAS inhibitors could eventually play a valuable role in treating this disease.
None.
Ethical policy
Non applicable.
Availability of data and materials
All data generated or analysed during this study are included in this publication.
Author contributions
Reham Gholam contributed to design of the work, data collection, and drafting the article. Muhammad Khalilzad was devoted to critical revision and final submission of the article.
Competing interests
The authors declare no competing interests.
Funding
None.
- Aden D, Sureka N, Zaheer S, Chaurasia JK, Zaheer S: Metabolic Reprogramming in Cancer: Implications for Immunosuppressive Microenvironment. Immunology 2025, 174(1): 30-72.
- Nong S, Han X, Xiang Y, Qian Y, Wei Y, Zhang T, Tian K, Shen K, Yang J, Ma X: Metabolic reprogramming in cancer: Mechanisms and therapeutics. MedComm (2020) 2023, 4(2): e218.
- Peixoto C, Martins M: Kidney Cancer Biomarker Selection Using Regularized Survival Models. Cells 2022, 11(15): 2311.
- Siddiqi A, Rani M, Bansal P, Rizvi MMA: Renal cell carcinoma management: A step to nano-chemoprevention. Life Sci 2022, 308: 120922.
- Hu SL, Chang A, Perazella MA, Okusa MD, Jaimes EA, Weiss RH: The Nephrologist's Tumor: Basic Biology and Management of Renal Cell Carcinoma. J Am Soc Nephrol 2016, 27(8): 2227-2237.
- Szymanski Ł, Helbrecht I, Fiedorowicz M, Matak D, Bartnik E, Golik P, Szczylik C, Czarnecka AM: [Cancer stem cells in renal carcinoma]. Postepy Biochem 2019, 65(2):95-102.
- Barata PC, Rini BI: Treatment of renal cell carcinoma: Current status and future directions. CA Cancer J Clin 2017, 67(6): 507-524.
- Motzer RJ, Powles T, Burotto M, Escudier B, Bourlon MT, Shah AY, Suárez C, Hamzaj A, Porta C, Hocking CM et al: Nivolumab plus cabozantinib versus sunitinib in first-line treatment for advanced renal cell carcinoma (CheckMate 9ER): long-term follow-up results from an open-label, randomised, phase 3 trial. Lancet Oncol 2022, 23(7): 888-898.
- Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, Pouliot F, Alekseev B, Soulières D, Melichar B et al: Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med 2019, 380(12): 1116-1127.
- Faubert B, Solmonson A: Metabolic reprogramming and cancer progression. Science 2020, 368(6487): eaaw5473.
- Warburg O: On the origin of cancer cells. Science 1956, 123(3191): 309-314.
- Wettersten HI, Aboud OA, Lara PN Jr, Weiss RH: Metabolic reprogramming in clear cell renal cell carcinoma. Nat Rev Nephrol 2017, 13(7): 410-419.
- Cassim S, Pouyssegur J: Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int J Mol Sci 2019, 21(1): 157.
- Jonasch E, Walker CL, Rathmell WK: Clear cell renal cell carcinoma ontogeny and mechanisms of lethality. Nat Rev Nephrol 2021, 17(4): 245-261.
- Hakimi AA, Reznik E, Lee CH, Creighton CJ, Brannon AR, Luna A, Aksoy BA, Liu EM, Shen R, Lee W et al: An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 2016, 29(1): 104-116.
- Fu Q, Xu L, Wang Y, Jiang Q, Liu Z, Zhang J, Zhou Q, Zeng H, Tong S, Wang T et al: Tumor-associated Macrophage-derived Interleukin-23 Interlinks Kidney Cancer Glutamine Addiction with Immune Evasion. Eur Urol 2019, 75(5): 752-763.
- Linehan WM, Schmidt LS, Crooks DR, Wei D, Srinivasan R, Lang M: The Metabolic Basis of Kidney Cancer. Cancer Discov 2019, 9(8): 1006-1021.
- Chitrakar A, Budda SA, Henderson JG, Axtell RC, Zenewicz LA: E3 Ubiquitin Ligase Von Hippel-Lindau Protein Promotes Th17 Differentiation. J Immunol 2020, 205(4): 1009-1023.
- Chappell JC, Payne LB, Rathmell WK: Hypoxia, angiogenesis, and metabolism in the hereditary kidney cancers. J Clin Invest 2019, 129(2): 442-451.
- Lee P, Chandel NS: Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol 2020, 21(5): 268-283.
- Al Tameemi W, Dale TP, Al-Jumaily RMK, Forsyth NR: Hypoxia-Modified Cancer Cell Metabolism. Front Cell Dev Biol 2019, 7: 4.
- Semenza GL: HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest 2013, 123(9): 3664-3671.
- Choudhry H, Harris AL: Advances in Hypoxia-Inducible Factor Biology. Cell Metab 2018, 27(2): 281-298.
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ et al: Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292(5516): 468-472.
- Motzer RJ, Banchereau R, Hamidi H, Powles T, McDermott D, Atkins MB, Escudier B, Liu LF, Leng N, Abbas AR et al: Molecular Subsets in Renal Cancer Determine Outcome to Checkpoint and Angiogenesis Blockade. Cancer Cell 2020, 38(6): 803-817.e804.
- Schönenberger D, Harlander S, Rajski M, Jacobs RA, Lundby AK, Adlesic M, Hejhal T, Wild PJ, Lundby C, Frew IJ: Formation of Renal Cysts and Tumors in Vhl/Trp53-Deficient Mice Requires HIF1α and HIF2α. Cancer Res 2016, 76(7): 2025-2036.
- LaGory EL, Wu C, Taniguchi CM, Ding CC, Chi JT, von Eyben R, Scott DA, Richardson AD, Giaccia AJ: Suppression of PGC-1α Is Critical for Reprogramming Oxidative Metabolism in Renal Cell Carcinoma. Cell Rep 2015, 12(1): 116-127.
- Dibble CC, Cantley LC: Regulation of mTORC1 by PI3K signaling. Trends Cell Biol 2015, 25(9): 545-555.
- Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C: PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A 2008, 105(35): 13057-13062.
- Yang P, Cornejo KM, Sadow PM, Cheng L, Wang M, Xiao Y, Jiang Z, Oliva E, Jozwiak S, Nussbaum RL et al: Renal cell carcinoma in tuberous sclerosis complex. Am J Surg Pathol 2014, 38(7): 895-909.
- Yu HB, Kielczewska A, Rozek A, Takenaka S, Li Y, Thorson L, Hancock REW, Guarna MM, North JR, Foster LJ et al: Sequestosome-1/p62 is the key intracellular target of innate defense regulator peptide. J Biol Chem 2009, 284(52): 36007-36011.
- Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S et al: Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 2010, 39(2): 171-183.
- Ivan M, Huang X: miR-210: fine-tuning the hypoxic response. Adv Exp Med Biol 2014, 772: 205-227.
- Schödel J, Grampp S, Maher ER, Moch H, Ratcliffe PJ, Russo P, Mole DR: Hypoxia, Hypoxia-inducible Transcription Factors, and Renal Cancer. Eur Urol 2016, 69(4): 646-657.
- Kondo K, Kim WY, Lechpammer M, Kaelin WG Jr: Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol 2003, 1(3): E83.
- Courtney KD, Ma Y, Diaz de Leon A, Christie A, Xie Z: HIF-2 Complex Dissociation, Target Inhibition, and Acquired Resistance with PT2385, a First-in-Class HIF-2 Inhibitor, in Patients with Clear Cell Renal Cell Carcinoma. Clin Cancer Res 2020, 26(4): 793-803.
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV: HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006, 3(3): 177-185.
- Infantino V, Santarsiero A: Cancer Cell Metabolism in Hypoxia: Role of HIF-1 as Key Regulator and Therapeutic Target. Int J Mol Sci 2021, 22(11): 5703.
- Xie H, Simon MC: Oxygen availability and metabolic reprogramming in cancer. J Biol Chem 2017, 292(41): 16825-16832.
- Nakazawa MS, Keith B, Simon MC: Oxygen availability and metabolic adaptations. Nat Rev Cancer 2016, 16(10): 663-673.
- Liu C, Li H, Huang H, Zheng P, Li Z: The Correlation of HK2 Gene Expression with the Occurrence, Immune Cell Infiltration, and Prognosis of Renal Cell Carcinoma. Dis Markers 2022, 2022: 1452861.
- Adeva-Andany MM, Pérez-Felpete N, Fernández-Fernández C, Donapetry-García C, Pazos-García C: Liver glucose metabolism in humans. Biosci Rep 2016, 36(6): e00416.
- Rui L: Energy metabolism in the liver. Compr Physiol 2014, 4(1): 177-197.
- Han HS, Kang G, Kim JS, Choi BH, Koo SH: Regulation of glucose metabolism from a liver-centric perspective. Exp Mol Med 2016, 48(3): e218.
- Wanders RJ, Vreken P, Ferdinandusse S, Jansen GA, Waterham HR, van Roermund CW, Van Grunsven EG: Peroxisomal fatty acid alpha- and beta-oxidation in humans: enzymology, peroxisomal metabolite transporters and peroxisomal diseases. Biochem Soc Trans 2001, 29(Pt 2): 250-267.
- Nogueira V, Hay N: Molecular pathways: reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin Cancer Res 2013, 19(16): 4309-4314.
- Aykin-Burns N, Ahmad IM, Zhu Y, Oberley LW, Spitz DR: Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem J 2009, 418(1): 29-37.
- Jiang P, Du W, Wu M: Regulation of the pentose phosphate pathway in cancer. Protein Cell 2014, 5(8): 592-602.
- Chakraborty S, Balan M, Sabarwal A, Choueiri TK, Pal S: Metabolic reprogramming in renal cancer: Events of a metabolic disease. Biochim Biophys Acta Rev Cancer 2021, 1876(1): 188559.
- Massari F, Ciccarese C, Santoni M, Brunelli M, Piva F, Modena A, Bimbatti D, Fantinel E, Santini D, Cheng L et al: Metabolic alterations in renal cell carcinoma. Cancer Treat Rev 2015, 41(9): 767-776.
- Wettersten HI, Hakimi AA, Morin D, Bianchi C, Johnstone ME, Donohoe DR, Trott JF, Aboud OA, Stirdivant S, Neri B et al: Grade-Dependent Metabolic Reprogramming in Kidney Cancer Revealed by Combined Proteomics and Metabolomics Analysis. Cancer Res 2015, 75(12): 2541-2552.
- Simonnet H, Alazard N, Pfeiffer K, Gallou C, Béroud C, Demont J, Bouvier R, Schägger H, Godinot C: Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis 2002, 23(5): 759-768.
- Yu Y, Yu Q, Zhang X: Allosteric inhibition of HIF-2α as a novel therapy for clear cell renal cell carcinoma. Drug Discov Today 2019, 24(12): 2332-2340.
- Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, McAfoos T, Morlacchi P, Ackroyd J: An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 2018, 24(7): 1036-1046.
- Zhu J, Thompson CB: Metabolic regulation of cell growth and proliferation. Nat Rev Mol Cell Biol 2019, 20(7): 436-450.
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, Morrison SJ: Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527(7577): 186-191.
- Hensley CT, Wasti AT, DeBerardinis RJ: Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest 2013, 123(9): 3678-3684.
- Yoo HC, Park SJ, Nam M, Kang J, Kim K, Yeo JH, Kim JK, Heo Y, Lee HS, Lee MY et al: A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab 2020, 31(2): 267-283.e212.
- Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, Cho SH, Paik Y, Wang Q, Zhang S et al: Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J Clin Invest 2021, 131(4): e140100.
- Pavlova NN, Zhu J, Thompson CB: The hallmarks of cancer metabolism: Still emerging. Cell Metab 2022, 34(3): 355-377.
- Lobo C, Ruiz-Bellido MA, Aledo JC, Márquez J, Núñez De Castro I, Alonso FJ: Inhibition of glutaminase expression by antisense mRNA decreases growth and tumourigenicity of tumour cells. Biochem J 2000, 348 Pt 2(Pt 2): 257-261.
- Wise DR, Thompson CB: Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 2010, 35(8): 427-433.
- Onishi Y, Hiraiwa M, Kamada H, Iezaki T, Yamada T, Kaneda K, Hinoi E: Hypoxia affects Slc7a5 expression through HIF-2α in differentiated neuronal cells. FEBS Open Bio 2019, 9(2): 241-247.
- Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ: Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res 2009, 69(20): 7986-7993.
- Wettersten HI: Reprogramming of Metabolism in Kidney Cancer. Semin Nephrol 2020, 40(1): 2-13.
- Fiore A, Murray PJ: Tryptophan and indole metabolism in immune regulation. Curr Opin Immunol 2021, 70: 7-14.
- Platten M, Nollen EAA, Röhrig UF: Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov 2019, 18(5): 379-401.
- Riesenberg R, Weiler C, Spring O, Eder M, Buchner A, Popp T, Castro M, Kammerer R, Takikawa O, Hatz RA et al: Expression of indoleamine 2,3-dioxygenase in tumor endothelial cells correlates with long-term survival of patients with renal cell carcinoma. Clin Cancer Res 2007, 13(23): 6993-7002.
- Trott JF, Kim J, Abu Aboud O, Wettersten H, Stewart B, Berryhill G, Uzal F, Hovey RC, Chen CH, Anderson K et al: Inhibiting tryptophan metabolism enhances interferon therapy in kidney cancer. Oncotarget 2016, 7(41): 66540-66557.
- Tang D, Yue L, Yao R, Zhou L, Yang Y, Lu L, Gao W: P53 prevent tumor invasion and metastasis by down-regulating IDO in lung cancer. Oncotarget 2017, 8(33): 54548-54557.
- Yao S, Janku F, Subbiah V: Phase 1 trial of ADI-PEG20 plus cisplatin in patients with pretreated metastatic melanoma or other advanced solid malignancies. Br J Cancer 2021, 124(9): 1533-1539.
- Heravi G, Yazdanpanah O, Podgorski I, Matherly LH, Liu W: Lipid metabolism reprogramming in renal cell carcinoma. Cancer Metastasis Rev 2022, 41(1): 17-31.
- Ganti S, Taylor SL, Abu Aboud O, Yang J, Evans C, Osier MV, Alexander DC, Kim K, Weiss RH: Kidney tumor biomarkers revealed by simultaneous multiple matrix metabolomics analysis. Cancer Res 2012, 72(14): 3471-3479.
- Gebhard RL, Clayman RV, Prigge WF, Figenshau R, Staley NA, Reesey C, Bear A: Abnormal cholesterol metabolism in renal clear cell carcinoma. J Lipid Res 1987, 28(10): 1177-1184.
- Saito K, Arai E, Maekawa K, Ishikawa M, Fujimoto H, Taguchi R, Matsumoto K, Kanai Y, Saito Y: Lipidomic Signatures and Associated Transcriptomic Profiles of Clear Cell Renal Cell Carcinoma. Sci Rep 2016, 6: 28932.
- Wu G, Wang Q, Xu Y, Li J, Zhang H, Qi G, Xia Q: Targeting the transcription factor receptor LXR to treat clear cell renal cell carcinoma: agonist or inverse agonist? Cell Death Dis 2019, 10(6): 416.
- Zou Y, Palte MJ, Deik AA, Li H, Eaton JK: A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun 2019, 10(1): 1617.
- Du W, Zhang L, Brett-Morris A, Aguila B, Kerner J, Hoppel CL, Puchowicz M, Serra D, Herrero L: HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat Commun 2017, 8(1): 1769.
- Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB et al: Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 2017, 13(1): 81-90.
- Chen X, Li J, Kang R, Klionsky DJ: Ferroptosis: machinery and regulation. Autophagy 2021, 17(9): 2054-2081.
- Huang C, Freter C: Lipid metabolism, apoptosis and cancer therapy. Int J Mol Sci 2015, 16(1): 924-949.
- Hanahan D, Weinberg RA: Hallmarks of cancer: the next generation. Cell 2011, 144(5): 646-674.
- Hsieh JJ, Le VH, Oyama T, Ricketts CJ, Ho TH, Cheng EH: Chromosome 3p Loss-Orchestrated VHL, HIF, and Epigenetic Deregulation in Clear Cell Renal Cell Carcinoma. J Clin Oncol 2018, 36(36): Jco2018792549.
- Keith B, Johnson RS, Simon MC: HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer 2011, 12(1): 9-22.
- Roskoski R Jr.: Vascular endothelial growth factor (VEGF) and VEGF receptor inhibitors in the treatment of renal cell carcinomas. Pharmacol Res 2017, 120: 116-132.
- Keefe D, Bowen J, Gibson R, Tan T, Okera M, Stringer A: Noncardiac vascular toxicities of vascular endothelial growth factor inhibitors in advanced cancer: a review. Oncologist 2011, 16(4): 432-444.
- Cassim S, Raymond VA, Dehbidi-Assadzadeh L, Lapierre P, Bilodeau M: Metabolic reprogramming enables hepatocarcinoma cells to efficiently adapt and survive to a nutrient-restricted microenvironment. Cell Cycle 2018, 17(7): 903-916.
- Abdel-Wahab AF, Mahmoud W, Al-Harizy RM: Targeting glucose metabolism to suppress cancer progression: prospective of anti-glycolytic cancer therapy. Pharmacol Res 2019, 150: 104511.
- DeBerardinis RJ, Keshari KR: Metabolic analysis as a driver for discovery, diagnosis, and therapy. Cell 2022, 185(15): 2678-2689.
- Kaelin WG Jr, Ratcliffe PJ: Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 2008, 30(4): 393-402.
- Koehler AN: A complex task? Direct modulation of transcription factors with small molecules. Curr Opin Chem Biol 2010, 14(3): 331-340.
- Scheuermann TH, Li Q, Ma HW, Key J, Zhang L, Chen R, Garcia JA, Naidoo J, Longgood J, Frantz DE et al: Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat Chem Biol 2013, 9(4): 271-276.
- Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, Homayoun F, Ma Y, Patel N, Yell P et al: Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539(7627): 112-117.
- Courtney KD, Infante JR, Lam ET, Figlin RA, Rini BI, Brugarolas J, Zojwalla NJ, Lowe AM, Wang K, Wallace EM et al: Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2α Antagonist in Patients With Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J Clin Oncol 2018, 36(9): 867-874.
- Xu R, Wang K, Rizzi JP, Huang H, Grina JA, Schlachter ST, Wang B, Wehn PM, Yang H, Dixon DD et al: 3-[(1S,2S,3R)-2,3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzonitrile (PT2977), a Hypoxia-Inducible Factor 2α (HIF-2α) Inhibitor for the Treatment of Clear Cell Renal Cell Carcinoma. J Med Chem 2019, 62(15): 6876-6893.
- Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, Oudard S, Else T, Maranchie JK, Welsh SJ et al: Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N Engl J Med 2021, 385(22): 2036-2046.
- Choueiri TK, Bauer TM, Papadopoulos KP, Plimack ER: Inhibition of hypoxia-inducible factor-2α in renal cell carcinoma with belzutifan: a phase 1 trial and biomarker analysis. Nat Med 2021, 27(5): 802-805.
- Emberley E, Pan A, Chen J, Dang R, Gross M, Huang T, Li W, MacKinnon A, Singh D, Sotirovska N et al: The glutaminase inhibitor telaglenastat enhances the antitumor activity of signal transduction inhibitors everolimus and cabozantinib in models of renal cell carcinoma. PLoS One 2021, 16(11): e0259241.
- Delage B, Fennell DA, Nicholson L, McNeish I, Lemoine NR, Crook T, Szlosarek PW: Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int J Cancer 2010, 126(12): 2762-2772.
- Haines RJ, Pendleton LC, Eichler DC: Argininosuccinate synthase: at the center of arginine metabolism. Int J Biochem Mol Biol 2011, 2(1): 8-23.
- Hall PE, Lewis R, Syed N, Shaffer R, Evanson J, Ellis S, Williams M: A Phase I Study of Pegylated Arginine Deiminase (Pegargiminase), Cisplatin, and Pemetrexed in Argininosuccinate Synthetase 1-Deficient Recurrent High-grade Glioma. Clin Cancer Res 2019, 25(9): 2708-2716.
- Tomlinson BK, Thomson JA, Bomalaski JS, Diaz M, Akande T, Mahaffey N, Li T, Dutia MP, Kelly K, Gong IY et al: Phase I Trial of Arginine Deprivation Therapy with ADI-PEG 20 Plus Docetaxel in Patients with Advanced Malignant Solid Tumors. Clin Cancer Res 2015, 21(11): 2480-2486.
- Abou-Alfa GK, Qin S, Ryoo BY, Lu SN, Yen CJ, Feng YH, Lim HY, Izzo F, Colombo M, Sarker D et al: Phase III randomized study of second line ADI-PEG 20 plus best supportive care versus placebo plus best supportive care in patients with advanced hepatocellular carcinoma. Ann Oncol 2018, 29(6): 1402-1408.
- Jochems C, Fantini M, Fernando RI, Kwilas AR, Donahue RN, Lepone LM, Grenga I, Kim YS, Brechbiel MW, Gulley JL et al: The IDO1 selective inhibitor epacadostat enhances dendritic cell immunogenicity and lytic ability of tumor antigen-specific T cells. Oncotarget 2016, 7(25): 37762-37772.
- Naing A, Powderly JD: Exploring the safety, effect on the tumor microenvironment, and efficacy of itacitinib in combination with epacadostat or parsaclisib in advanced solid tumors: a phase I study. J Immunother Cancer 2022, 10(3): e004223.
- Nayak-Kapoor A, Hao Z, Sadek R, Dobbins R, Marshall L, Vahanian NN, Jay Ramsey W, Kennedy E, Mautino MR, Link CJ et al: Phase Ia study of the indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) in patients with recurrent advanced solid tumors. J Immunother Cancer 2018, 6(1): 61.
- Ma S, Suchomel J, Yanez E, Yost E, Liang X, Zhu R, Le H, Siebers N, Joas L, Morley R et al: Investigation of the absolute bioavailability and human mass balance of navoximod, a novel IDO1 inhibitor. Br J Clin Pharmacol 2019, 85(8): 1751-1760.
- Jung KH, LoRusso P, Burris H, Gordon M, Bang YJ, Hellmann MD: Phase I Study of the Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Navoximod (GDC-0919) Administered with PD-L1 Inhibitor (Atezolizumab) in Advanced Solid Tumors. Clin Cancer Res 2019, 25(11): 3220-3228.
- Angeles TS, Hudkins RL: Recent advances in targeting the fatty acid biosynthetic pathway using fatty acid synthase inhibitors. Expert Opin Drug Discov 2016, 11(12): 1187-1199.
- Horiguchi A, Asano T, Asano T, Ito K, Sumitomo M, Hayakawa M: Fatty acid synthase over expression is an indicator of tumor aggressiveness and poor prognosis in renal cell carcinoma. J Urol 2008, 180(3): 1137-1140.
- Horiguchi A, Asano T, Asano T, Ito K, Sumitomo M, Hayakawa M: Pharmacological inhibitor of fatty acid synthase suppresses growth and invasiveness of renal cancer cells. J Urol 2008, 180(2): 729-736.
- Wang J, Lin W, Li R, Cheng H, Sun S, Shao F, Yang Y, Zhang L, Feng X, Gao S et al: The Deubiquitinase USP13 Maintains Cancer Cell Stemness by Promoting FASN Stability in Small Cell Lung Cancer. Front Oncol 2022, 12: 899987.
Annals of urologic oncology
p-ISSN: 2617-7765, e-ISSN: 2617-7773
 Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.