
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
Basic Study on Gene Biology of Bladder Cancer Metastasis
Clara Joseph1
1Faculty of Health Sciences, Busitema University, Tororo, Uganda.
Correspondence: Clara Joseph (Faculty of Health Sciences, Busitema University, Tororo, Uganda; Email: clara32@runbox.com).
Annals of Urologic Oncology 2024, 7(4): 148-157. https://doi.org/10.32948/auo.2024.10.22
Received: 25 Sep 2024 | Accepted: 04 Nov 2024 | Published online: 13 Nov 2024
Key words bladder cancer, tumor metastasis, basic research, biology
Urothelium is the epithelial tissue that lines the entire urinary tract, including the renal pelvis, ureters, bladder, and proximal urethra. Bladder cancer is the most common tumor in the urothelium. The bladder serves as a storage organ for urine and acts as a barrier between urine and blood, and it is frequently exposed to various potential carcinogens [3]. Compared to other types of tumors, bladder cancer exhibits distinct characteristics of progression through various pathways that include Non-Muscle Invasive Bladder Cancer (NMIBC), accounting for 70-80% of bladder cancers, is typically of low grade, superficial, and papillary. Approximately 70% of these tumors recur, but only about 15% progress to muscle invasion. These tumors often harbor mutations in the HRAS gene (30-40%) and the Fibroblast Growth Factor Receptor (FGFR3) gene (~70%), indicating that the activation of RTK-Ras plays an early and crucial role in this pathway of tumorigenesis. Muscle-Invasive Bladder Cancer (MIBC), which accounts for 20-30% of bladder cancers, is characterized by high-grade muscle-invasive tumors. These tumors may either originate from flat carcinoma in situ (CIS) or present high-grade and invasive features from the onset of tumorigenesis. More than half of MIBC cases contain structural and functional defects in tumor suppressor factors such as p53 and/or retinoblastoma protein (RB), with over 50% progressing to local and distant metastasis [4]. In bladder cancer, MIBC (muscle-invasive subtype) is the major cause of bladder cancer-related deaths. The five-year survival rate for MIBC drops from less than 50% in the absence of metastasis to 5% in cases with distant metastasis, indicating that metastatic factors in MIBC are the primary contributors to bladder cancer deaths. Studying the biological processes of bladder cancer cell metastasis is of great value for identifying potential therapeutic targets and improving the prognosis of bladder cancer patients [5].
One crucial lesson from humanity’s long battle against disease is that only by deepening our understanding of disease mechanisms can we develop more effective treatment strategies. Currently, effective cancer treatments are based on a deeper understanding of tumor biology, with the development and use of targeted therapies serving as a prime example of this approach. Among these, a notable example is the development of Imatinib (Gleevec). Through the exploration of disease mechanisms, researchers ultimately discovered that the BCR-ABL fusion gene is the cause of chronic myeloid leukemia (CML). Through ongoing design and refinement of the drug molecules, researchers successfully developed Imatinib mesylate, transforming CML into a manageable chronic disease. Similarly, by gaining a thorough understanding of the mechanisms that hinders the immune system’s ability to eliminate tumor cells, scientists developed immune checkpoint inhibitors, greatly enhancing the immune system's ability to target and destroy tumors. For example, antibodies and small molecule inhibitors based on PD-1/PD-L1, as well as Chimeric Antigen Receptor T-cell Immunotherapy (CAR-T), were developed. In conclusion, understanding the mechanisms behind tumor metastasis is essential for developing more effective strategies to combat it. This review will explore bladder cancer metastasis from a researcher's perspective, examining various key aspects of this complex process.
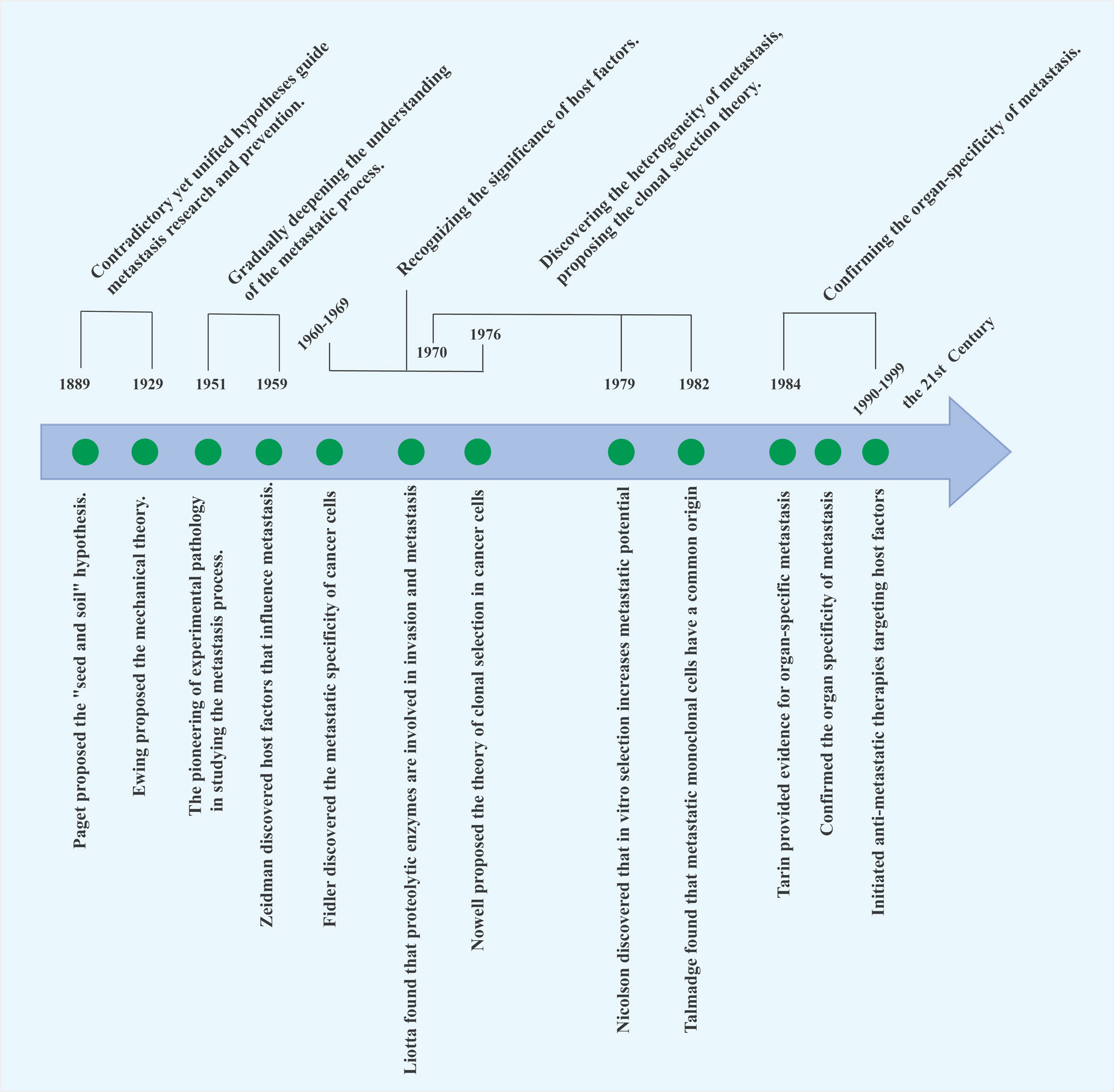 Figure 1. Timeline of tumor metastasis research. This timeline represents a historical overview of the significant milestones and discoveries in the field of tumor metastasis research, highlighting key breakthroughs that have shaped our understanding of the metastatic process.
Figure 1. Timeline of tumor metastasis research. This timeline represents a historical overview of the significant milestones and discoveries in the field of tumor metastasis research, highlighting key breakthroughs that have shaped our understanding of the metastatic process.
There are mainly two types of models: syngeneic transplantation models and transgenic mouse models. The former refers to the inoculation of tumor cells or tissues from the same species, such as mice, into the subcutaneous tissue, vascular system, or primary tumor site, to observe the tumor growth, progression, metastasis, and other processes. In this model, both the graft and the host belong to the same species, and the host retains normal immune function. Another method is carcinogen-induced models In bladder cancer research, the most common spontaneous tumor model is the BBN-induced mouse bladder cancer model. However, the heterogeneity of the cancer phenotype makes it difficult to use in molecular research or preclinical studies, and it rarely forms metastases [6, 7].
Transgenic mouse models primarily refer to models that develop tumors spontaneously through gene modification, leading to progression and metastasis. For instance, the MMTV-polyoma virus middle T antigen (PyVmt) transgenic mouse is a model often used in breast cancer research. Through gene modification and exposure to carcinogens, tumors naturally develop in specific organs or sites within the mouse, subsequently metastasizing to distant organs. Transgenic mice maintain relatively intact immune system functions, making the research results more reflective of real-world scenarios. Compared to other cancers, the availability of bladder cancer models is somewhat limited, with the representative nature of genetically engineered mouse (GEM) models being notably deficient [8]. Transgenic spontaneous bladder cancer models, for example, involve the expression of oncogenes in the urinary epithelial cells of mice through genetic modification. Germline models, such as the construction of P53 knockout mice or mice with conditional knockout of tumor suppressor genes, are also included. However, these models have the following disadvantages: 1. Limited selection of genes that are specifically expressed in the bladder; 2. The phenotypes are relatively mild; 3. There are very few models that develop invasive bladder cancer; 4. Metastasis is rarely observed [9-11].
Xenograft models
Primarily, human tumor cells or fragments are implanted into immunodeficient experimental animals. It is clear that one significant disadvantage of this model is the inability to observe the role of the immune system in tumor progression and metastasis [12]. Nevertheless, the xenograft model has been proven to be valuable for understanding cancer biology and metastasis due to its relatively short duration and low cost, making significant contributions to the study of tumor metastasis. Furthermore, the application of orthotopic implantation techniques more closely resembles the complex tumor microenvironment and growth, providing a more comprehensive simulation of the entire metastasis process [8]. It is worth noting that the orthotopic bladder cancer model has not been widely used due to the thin bladder wall and the large volume of the free portion. Gabri van der Pluijm and colleagues utilized poly-L-lysine to damage the bladder wall while retaining perfusion of luciferase-labeled tumor cells, allowing for a comprehensive and efficient simulation of the entire process of bladder cancer progression and distant metastasis. This approach offers higher cost-effectiveness and practicality, making it an ideal preclinical model [13]. Furthermore, the PDX (Patient-Derived Xenograft) mouse model, which involves implanting a patient's tumor into recipient mice, also falls under the category of heterologous models. The tumor in PDX mice is directly derived from the patient, providing a clear advantage in selecting new drugs after the tumor develops drug resistance. An increasing number of PDX models are rapidly replacing long-established traditional cell lines, becoming the preferred model for conducting basic and translational preclinical research [14].
Tumor metastasis suppressor genes
The definition of metastasis suppressor genes highlights their important role in inhibiting the formation of metastases. The earliest successful technique used for the screening of metastasis suppressor genes (MSG) was the MMCT method. Specifically, this involves inhibiting the mitosis of growing cells, allowing chromosomes to drift naturally, and then encapsulating the drifting chromosomes into new micronuclei. Subsequently, employing methods such as altering their chemical properties, differential centrifugation, and filtration, microcells containing a single chromosome were selected. These microcells are then fused with tumor cells to form new hybrid cells, in order to screen for MSG. Although the efficiency is low and the steps are complex, through the persistent efforts of previous researchers, genes such as BRMS1 have been identified [16]. With the advancement of high-throughput technologies such as chips and sequencing, a series of MSGs have been identified and validated (Table 1). Except for a few genes like RhoGDI2 and CDH1, most genes in the table lack direct experimental evidence for their role in inhibiting bladder cancer metastasis. By elucidating the roles and mechanisms of these genes in bladder cancer metastasis, it is hoped that potential therapeutic targets with application value can be discovered.
Oncogene promoting tumor metastasis
Receptor tyrosine kinase (RTK). Receptor Tyrosine Kinases (RTKs) are a family of cell surface receptors, with 58 known RTKs in humans, which can be divided into 20 subfamilies [53]. RTKs serve as receptors for growth factors, hormones, cytokines, neurotrophic factors, and other extracellular signaling molecules. RTKs mediate key signaling pathways involved in cell proliferation, differentiation, survival, and cell migration, where the activation mutation or overexpression of these molecules plays an important role in tumor development and progression. Considering EGFR as an example, when the ligand binds to it, the bound EGFR receptor undergoes a conformational change in its extracellular domain, leading to dimerization with other receptors of the same or similar family members. This dimerization results in the mutual phosphorylation of specific sites on EGFR, thereby activating the receptor. These phosphorylated sites serve as docking points for various scaffolding proteins or kinases originally located in the cytoplasm, facilitating their translocation to the membrane and thereby activating a series of downstream pathways that regulate cell behavior. These scaffolding proteins include PKC, PI3K, RAS, SRC, ABL, PAK and STAT5 [53, 54].
EGFR. More than 50% of tumors exhibit high expression of EGFR, and its expression level is directly related to tumor grade, stage, and survival rates. In muscle-invasive bladder cancer (MIBC), overexpression of EGFR is associated with lower tumor-specific survival. The relationship between EGFR and the invasion and metastasis of bladder cancer is also reflected in preclinical models: compared to 253J cells, 253JBV cells (which are highly metastatic cells screened after continuous passage in mice) exhibit overexpression of EGFR [55, 56]. Similarly, other experiments have confirmed that EGFR promotes metastasis by enhancing the proliferation, angiogenesis, and invasion of bladder cancer cells. EGFR family consists of four members: in addition to the previously mentioned EGFR, there are human EGFR 2, human EGFR 3, and human EGFR 4. EGFR2 cannot form homodimers; instead, it functions by forming heterodimers with other EGFR members, which then transmit signals downstream to regulate cellular behavior [57]. Expression of EGFR2 positively correlated with the metastatic capacity of MIBC [58]. Although there are studies suggesting that EGFR3 and EGFR4 may play significant roles in the progression of bladder cancer, there is no direct evidence demonstrating that EGFR3/4 directly promotes bladder cancer metastasis. EGFR can promote bladder cancer metastasis by regulating the EMT (epithelial-mesenchymal transition) process. Preclinical studies on other cancer models have identified that EGFR can promote EMT through STAT3 signaling, suggesting that high levels of EGFR expression may likely contribute to EMT in basal-like bladder cancer [59].
Matrix metalloproteinases (MMPs). Matrix metalloproteinases (MMPs) are a family of proteolytic enzymes that function extracellularly by altering the cellular microenvironment. Their substrates include basement membranes, extracellular matrix (ECM) molecules, and various extracellular cytokines, indicating that MMPs play a central role in normal physiological processes, benign diseases, and malignant tumors [16]. MMPs are crucial for tumor-induced angiogenesis, tumor invasion, and the establishment of metastatic foci at secondary sites [60]. Tumor cells must detach from the primary site, degrade the extracellular matrix (ECM), and invade the stroma as a prerequisite to enter the bloodstream and colonize at distant sites. Moreover, at the secondary sites, tumor cells must induce and establish a blood supply to create conditions favorable for proliferation [61, 62]. Although invasion and angiogenesis are very different processes, both invasion and angiogenesis require proteases such as MMP-9 to alter the extracellular matrix and basement membranes [63]. Studies have shown that the mRNA expression levels of MMP2 and MMP9 are higher in muscle-invasive bladder cancer (MIBC) than in non-muscle-invasive bladder cancer (NMIBC), and their high expression is negatively correlated with bladder cancer survival rates, indicating prognostic value in MIBC [64, 65]. Moreover, their roles in the proteolytic processing of various growth factors, growth factor receptors, and cytokines have been recognized. In a different mouse model, after intracardiac injection of bladder cancer cells, continuous retrieval of bone metastatic foci revealed increasing levels of MT1-MMP, MT2-MMP, MMP-9 and TIMP-2, which were associated with metastatic potential. The use of MMP-2 and MMP-9 gene knockout mice further reinforced the importance of these genes in metastasis [67, 68]. Notably, in MMP-9 gene knockout mice, there was a significant reduction in spontaneous metastasis compared to wild-type mice, using different types of tumors [66].
RHO family genes. Small GTP-binding protein Rho and its most typical downstream effector, Rho-associated serine/threonine protein kinase (ROCK), are involved in actin cytoskeletal organization and are associated with the pathogenesis and progression of several human cancers [69, 70]. Members of the Rho subfamily of small GTPases are involved in regulating various cellular processes, including microfilament organization, cell-to-cell contact, and malignant transformation of cells [70]. Indeed, these events are also interrelated. Specifically, the Rho subfamily regulates the formation of intracellular stress fibers and focal adhesions. The Rac subfamily regulates the formation of lamellipodia and membrane ruffles, while the Cdc42 subfamily regulates the formation of filopodia [71, 72]. Lamellipodia and filopodia appear on the leading edge of motile cells, while retraction occurs on the trailing edge [73]. These changes are the basis of cell migration. Cancer cell migration is central to the metastatic process. The molecular alterations described above, resulting in corresponding changes in cell migration ability, are crucial for the in vivo metastasis of tumor cells. Rho proteins act as molecular switches inside cells, transmitting extracellular stimuli signals to the actin cytoskeleton and nucleus, regulating cell migration and malignant transformation. The rho/rock signaling pathway is involved in the regulation of the cytoskeleton, thereby affecting various behaviors such as cell migration and invasion, promoting tumor metastasis. By regulating the RhoA/ROCK signaling pathway, MALAT1 promotes the metastasis of osteosarcoma [74]. AFAP1-AS1 may promote the metastasis of nasopharyngeal carcinoma cells by regulating tumor cell adhesion and migration through the RhoA/Rac2 signaling pathway [75]. In the previous section on tumor suppressor genes, it is mentioned that one of the key tumor suppressor genes identified in bladder cancer is RhoGDI2, whose full name is Rho GDP dissociation inhibitor 2. This gene exerts its function by binding to Rho family members and inhibiting their activity. Researchers have found that reactivation of RhoGDI2 can effectively prevent the occurrence of bladder cancer invasion and metastasis events [76]. These results indicate that some members of the Rho family are key factors in promoting bladder cancer metastasis. Additionally, MMP-2, as a downstream molecule of RhoGDI2, is positively regulated by it and plays a role in the invasion of bladder cancer into surrounding tissues and the metastatic process [77].
|
Table 1. Tumor metastasis suppressor genes. |
||
|
Gene |
As evidence for metastasis suppressor genes |
Reference |
|
CDH1 |
Cell-cell/cell-matrix adhesion |
[17] |
|
CDH11 |
Knockdown inhibits EMT and tumor cell stemness. |
[18, 19] |
|
CRSP3 |
Overexpression exogenously can inhibit metastasis. |
[20] |
|
KISS1/KISS1R |
Exogenous overexpression of KISS1 allows cells to disseminate, but they do not proliferate in the secondary organs. |
[20, 21] |
|
KLF17 |
Overexpression inhibits metastasis; knockdown promotes EMT transformation. |
[22] |
|
MAK7 |
Regulating the MAPK pathway affects metastasis. |
[23] |
|
AKAP12 |
Functioning through JN and/or Raf/MEK/ERK. |
[24] |
|
BRMS1 |
In experimental metastasis analysis, reduced tumor metastasis rate and size. |
[25] |
|
Caspase 8 |
Induced apoptosis/anoikis. |
[26] |
|
CD44 |
Cell-cell/cell-matrix adhesion. |
[27, 28] |
|
Claudin-4 |
Inhibiting anchorage-independent growth processes. |
[29] |
|
CTGF |
Lung metastatic lesions size reduction by 15%-25%. |
[30, 31] |
|
DCC |
Cell-cell/cell-matrix adhesion; Apoptosis. |
[32] |
|
DLC1 |
After re-expression, significant inhibition of tumor metastasis. |
[33, 34] |
|
DRG1 |
Overexpression of Drg-1 in metastatic colon cancer cells reduces in vitro invasion through Matrigel and inhibits liver metastasis in nude mice. |
[35, 36]
|
|
GAS1 |
GAS1 inhibits the glycolytic process, thereby suppressing tumor metastasis. |
[18]
|
|
Gelsolin |
Affecting the cytoskeleton affects metastasis. |
[37, 38] |
|
KAI-1 |
Binding to DARC on the surface of vascular endothelial cells induces tumor cell growth arrest. |
[39]
|
|
LSD1 |
The influence on the cytoskeleton affects metastasis. |
[40] |
|
MAP2K4 |
Constitutively active MAP2K4 increases tumor size and the number of circulating tumor cells in the blood and bone marrow. |
[41]
|
|
MKK-4 |
Overexpression subsequently inhibits the metastasis process. |
[42] |
|
MKK-7 |
Overexpression suppresses the metastatic process. |
[43] |
|
Nm23 |
Inhibition of metastatic clone growth. |
[44] |
|
OGR-1 |
Inhibition of the metastasis process. |
[45] |
|
PEBP1 |
Referred to as Raf kinase inhibitor protein (RKIP). |
[46] |
|
RhoGDI2 |
Reduce tumor metastasis rate and size; it is an important target of the anti-angiogenic compound apatinib. |
[47]
|
|
RKIP |
Regulate the expression of angiogenic genes; regulate the checkpoint of the spindle in the cell cycle. |
[48]
|
|
RRM1 |
Inhibit tumor metastasis by positively regulating PTEN. |
[49] |
|
SMAD7 |
Overexpression of Smad7 delays the establishment and growth of a mouse model of melanoma bone metastasis. |
[50]
|
|
SSeCKS |
Reducing the rate and size of tumor metastasis. |
[51] |
|
TXNIP |
Influencing metastasis by regulating cellular redox homeostasis. |
[20, 52]
|
The whole-genome mRNA expression profiles of primary bladder cancer and circulating tumor cells (CTCs) in bladder cancer show that tumor cells in the bloodstream have higher expression levels of EMT markers, which is consistent with observations in other cancer models [83-85]. Notably, CTCs exhibit increased expression of SNAI1 (Snail), and conditional knockout of SNAI1 blocks the production and metastasis of CTCs. When exosomes from MIBC cell lines stimulate primary urothelial carcinoma cells (Figure 2), the urothelial cells undergo EMT, accompanied by enhanced cell migration abilities [86]. At the molecular level, EMT is characterized by the loss of E-cadherin and increased expression of several transcriptional repressors of E-cadherin. A significant feature of bladder cancer is its development along two seemingly distinct pathways at the phenotypic and molecular levels. Researchers have conducted clustering analysis on the expression of EMT markers (E-cadherin, Zeb-1, and Zeb-2) and the "dual-track" developmental pathways exhibited by bladder cancer. The results confirmed that the expression of Zeb-1 and Zeb-2 is highly enriched in MIBC, supporting the concept that EMT may serve as a basis for the invasion and metastasis of bladder cancer. These findings indicate that EMT plays a crucial regulatory role in the invasion/metastasis process of various tumors, including bladder cancer, and it is also a major characteristic of the entire cancer biology.
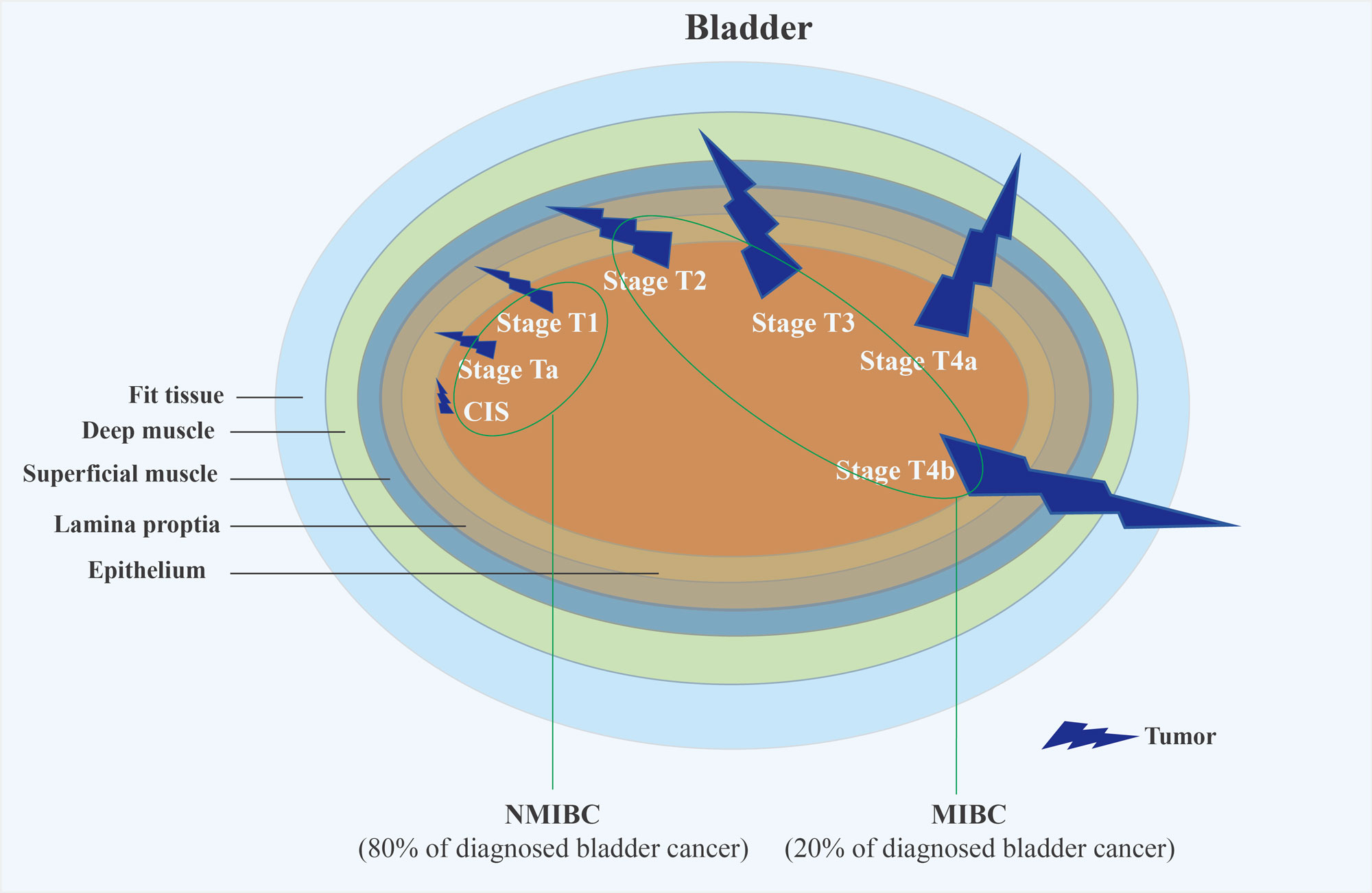 Figure 2. The TNM staging system for bladder cancer is a standardized method used by healthcare professionals to classify and categorize the extent of the cancer based on three key components: Tumor (T), Nodes (N), and Metastasis (M). This system helps in determining the stage of the cancer, which in turn influences treatment options and prognostic assessments.
Figure 2. The TNM staging system for bladder cancer is a standardized method used by healthcare professionals to classify and categorize the extent of the cancer based on three key components: Tumor (T), Nodes (N), and Metastasis (M). This system helps in determining the stage of the cancer, which in turn influences treatment options and prognostic assessments.
In the earlier chapters of this review, we introduced MSGs—metastasis suppressor genes. These genes are unified by their ability to inhibit metastasis. As researchers explore the mechanisms by which MSGs prevent metastasis, they have found that MSGs play a significant role in promoting the dormancy of disseminated tumor cells, which is one of their key functions [90]. RhoGDI2 has been defined as a gene that inhibits metastasis without affecting the proliferation of primary tumors. Its mechanism may involve inducing or maintaining tumor cells in a dormant state [91]. Evidence suggests that the metastasis suppressor factor RhoGDI2 inhibits the endothelin axis and interacts with macrophages in the microenvironment of micrometastasis, thereby suppressing metastatic growth.
Another MSG we mentioned earlier, KISS1, is a ligand for the G protein-coupled receptor (KISS1R) and exerts a strong inhibitory effect on metastasis across various tumor types, including breast cancer, melanoma, prostate cancer, ovarian cancer, and pancreatic cancer. Studies in these tumors suggest that the mechanism by which kiss1/kissR inhibits tumors may also involve mediating tumor cell dormancy [88]. Furthermore, the dormancy of tumors in target organs, while making metastasis appear inefficient, is not uncommon for cases where, years after the removal of the primary tumor, cells re-enter the cell cycle and proliferate under suitable conditions, leading to recurrence [88]. This indicates that research into this mechanism has potential clinical significance. In summary, the study of tumor dormancy poses a challenging issue. A deeper understanding of the mechanisms underlying this phenomenon will guide the development of new therapeutic strategies targeting tumor metastasis.
None.
Ethical policy
All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study. Approval from institutional ethical committee was taken.
Availability of data and materials
All data generated or analysed during this study are included in this publication.
Author contributions
CJ searched academic literature, wrote the draft manuscript, made the figures and submitted the final manuscript.
Competing interests
Authors report no conflict of interest.
Funding
None.
- Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y: Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduct Target Ther 2020, 5(1): 28-40.
- Lambert AW, Pattabiraman DR, Weinberg RA: Emerging Biological Principles of Metastasis. Cell 2017, 168(4): 670-691.
- Pardo JC, Ruiz de Porras V, Plaja A, Carrato C, Etxaniz O, Buisan O, Font A: Moving towards Personalized Medicine in Muscle-Invasive Bladder Cancer: Where Are We Now and Where Are We Going? Int J Mol Sci 2020, 21(17): 6271-282.
- Witjes JA, Bruins HM, Cathomas R, Compérat EM, Cowan NC, Gakis G, Hernández V, Linares Espinós E, Lorch A, Neuzillet Y, et al: European Association of Urology Guidelines on Muscle-invasive and Metastatic Bladder Cancer: Summary of the 2020 Guidelines. Eur Urol 2021, 79(1): 82-104.
- Tran L, Xiao JF, Agarwal N, Duex JE, Theodorescu D: Advances in bladder cancer biology and therapy. Nat Rev Cancer 2021, 21(2): 104-121.
- Kim SK, Yun SJ, Kim J, Lee OJ, Bae SC, Kim WJ: Identification of gene expression signature modulated by nicotinamide in a mouse bladder cancer model. PLoS One 2011, 6(10): e26131-e26152.
- Yamamoto S, Masui T, Murai T, Mori S, Oohara T, Makino S, Fukushima S, Tatematsu M: Frequent mutations of the p53 gene and infrequent H- and K-ras mutations in urinary bladder carcinomas of NON/Shi mice treated with N-butyl-N-(4-hydroxybutyl)nitrosamine. Carcinogenesis 1995, 16(10): 2363-2368.
- Kobayashi T, Owczarek TB, McKiernan JM, Abate-Shen C: Modelling bladder cancer in mice: opportunities and challenges. Nat Rev Cancer 2015, 15(1): 42-54.
- Ding J, Xu D, Pan C, Ye M, Kang J, Bai Q, Qi J: Current animal models of bladder cancer: Awareness of translatability (Review). Exp Ther Med 2014, 8(3): 691-699.
- DeGraff DJ, Robinson VL, Shah JB, Brandt WD, Sonpavde G, Kang Y, Liebert M, Wu XR, et al: Current preclinical models for the advancement of translational bladder cancer research. Mol Cancer Ther 2013, 12(2): 121-30.
- Ahmad I, Sansom OJ, Leung HY: Exploring molecular genetics of bladder cancer: lessons learned from mouse models. Dis Model Mech 2012, 5(3): 323-332.
- de Visser KE, Eichten A, Coussens LM: Paradoxical roles of the immune system during cancer development. Nat Rev Cancer 2006, 6(1): 24-37.
- van der Horst G, van Asten JJ, Figdor A, van den Hoogen C, Cheung H, Bevers RF, Pelger RC, van der Pluijm G: Real-time cancer cell tracking by bioluminescence in a preclinical model of human bladder cancer growth and metastasis. Eur Urol 2011, 60(2): 337-343.
- Dobrolecki LE, Airhart SD, Alferez DG, Aparicio S, Behbod F, Bentires-Alj M, Brisken C, Bult CJ, Cai S, Clarke RB, et al: Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Rev 2016, 35(4): 547-573.
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al: A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10(6): 515-527.
- Lu W, Kang Y: Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell 2019, 49(3): 361-374.
- Kim SA, Inamura K, Yamauchi M, Nishihara R, Mima K, Sukawa Y, Li T, Yasunari M, Morikawa T, Fitzgerald KC, et al: Loss of CDH1 (E-cadherin) expression is associated with infiltrative tumour growth and lymph node metastasis. Br J Cancer 2016, 114(2): 199-206.
- Yang Z, Yan C, Yu Z, He C, Li J, Li C, Yan M, Liu B, Wu Y, Zhu Z, et al: Downregulation of CDH11 Promotes Metastasis and Resistance to Paclitaxel in Gastric Cancer Cells. J Cancer 2021, 12(1): 65-75.
- Chen JH, Huang WC, Bamodu OA, Chang PM, Chao TY, Huang TH: Monospecific antibody targeting of CDH11 inhibits epithelial-to-mesenchymal transition and represses cancer stem cell-like phenotype by up-regulating miR-335 in metastatic breast cancer, in vitro and in vivo. BMC Cancer 2019, 19(1): 634-645.
- Goldberg SF, Miele ME, Hatta N, Takata M, Paquette-Straub C, Freedman LP, Welch DR: Melanoma metastasis suppression by chromosome 6: evidence for a pathway regulated by CRSP3 and TXNIP. Cancer Res 2003, 63(2): 432-440.
- Rinker-Schaeffer CW, O'Keefe JP, Welch DR, Theodorescu D: Metastasis suppressor proteins: discovery, molecular mechanisms, and clinical application. Clin Cancer Res 2006, 12(13): 3882-3889.
- Goldberg SF, Miele ME, Hatta N, Takata M, Paquette-Straub C, Freedman LP, Welch DR: Melanoma metastasis suppression by chromosome 6: evidence for a pathway regulated by CRSP3 and TXNIP. Cancer Res 2003, 63(2): 432-440.
- Yan J, Yang Q, Huang Q: Metastasis suppressor genes. Histol Histopathol 2013, 28(3): 285-292.
- Wu X, Wu T, Li K, Li Y, Hu TT, Wang WF, Qiang SJ, Xue SB, Liu WW: The Mechanism and Influence of AKAP12 in Different Cancers. Biomed Environ Sci 2018, 31(12): 927-932.
- Goriki A, Seiler R, Wyatt AW, Contreras-Sanz A, Bhat A, Matsubara A, Hayashi T, Black PC: Unravelling disparate roles of NOTCH in bladder cancer. Nat Rev Urol 2018, 15(6): 345-357.
- Stupack DG, Teitz T, Potter MD, Mikolon D, Houghton PJ, Kidd VJ, Lahti JM, Cheresh DA: Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature 2006, 439(7072): 95-99.
- Lopez JI, Camenisch TD, Stevens MV, Sands BJ, McDonald J, Schroeder JA: CD44 attenuates metastatic invasion during breast cancer progression. Cancer Res 2005, 65(15): 6755-6763.
- Lesley J, Hyman R, English N, Catterall JB, Turner GA: CD44 in inflammation and metastasis. Glycoconj J 1997, 14(5): 611-622.
- Michl P, Barth C, Buchholz M, Lerch MM, Rolke M, Holzmann KH, Menke A, Fensterer H, Giehl K, Löhr M, et al: Claudin-4 expression decreases invasiveness and metastatic potential of pancreatic cancer. Cancer Res 2003, 63(19): 6265-6271.
- Kemp CJ, Moore JM, Moser R, Bernard B, Teater M, Smith LE, Rabaia NA, Gurley KE, Guinney J, Busch SE, et al: CTCF haploinsufficiency destabilizes DNA methylation and predisposes to cancer. Cell Rep 2014, 7(4): 1020-1029.
- Zhang B, Zhang Y, Zou X, Chan AW, Zhang R, Lee TK, Liu H, Lau EY, Ho NP, Lai PB, et al: The CCCTC-binding factor (CTCF)-forkhead box protein M1 axis regulates tumour growth and metastasis in hepatocellular carcinoma. J Pathol 2017, 243(4): 418-430.
- Saito M, Yamaguchi A, Goi T, Tsuchiyama T, Nakagawara G, Urano T, Shiku H, Furukawa K: Expression of DCC protein in colorectal tumors and its relationship to tumor progression and metastasis. Oncology 1999, 56(2): 134-141.
- Barras D, Widmann C: GAP-independent functions of DLC1 in metastasis. Cancer Metastasis Rev 2014, 33(1): 87-100.
- Goodison S, Yuan J, Sloan D, Kim R, Li C, Popescu NC, Urquidi V: The RhoGAP protein DLC-1 functions as a metastasis suppressor in breast cancer cells. Cancer Res 2005, 65(14): 6042-6053.
- Baig RM, Sanders AJ, Kayani MA, Jiang WG: Association of Differentiation-Related Gene-1 (DRG1) with Breast Cancer Survival and in Vitro Impact of DRG1 Suppression. Cancers (Basel) 2012, 4(3): 658-672.
- Guan RJ, Ford HL, Fu Y, Li Y, Shaw LM, Pardee AB: Drg-1 as a differentiation-related, putative metastatic suppressor gene in human colon cancer. Cancer Res 2000, 60(3): 749-755.
- Stock AM, Klee F, Edlund K, Grinberg M, Hammad S, Marchan R, Cadenas C, Niggemann B, Zänker KS, Rahnenführer J, et al: Gelsolin Is Associated with Longer Metastasis-free Survival and Reduced Cell Migration in Estrogen Receptor-positive Breast Cancer. Anticancer Res 2015, 35(10): 5277-5285.
- Yuan X, Yu L, Li J, Xie G, Rong T, Zhang L, Chen J, Meng Q, Irving AT, Wang D, et al: ATF3 suppresses metastasis of bladder cancer by regulating gelsolin-mediated remodeling of the actin cytoskeleton. Cancer Res 2013, 73(12): 3625-3637.
- Dong JT, Lamb PW, Rinker-Schaeffer CW, Vukanovic J, Ichikawa T, Isaacs JT, Barrett JC: KAI1, a metastasis suppressor gene for prostate cancer on human chromosome 11p11.2. Science 1995, 268(5212): 884-896.
- Ketscher A, Jilg CA, Willmann D, Hummel B, Imhof A, Rüsseler V, Hölz S, Metzger E, Müller JM, Schüle R, et al: LSD1 controls metastasis of androgen-independent prostate cancer cells through PXN and LPAR6. Oncogenesis 2014, 3(10): e120.
- Pavese JM, Ogden IM, Voll EA, Huang X, Xu L, Jovanovic B, Bergan RC: Mitogen-activated protein kinase kinase 4 (MAP2K4) promotes human prostate cancer metastasis. PLoS One 2014, 9(7): e102289.
- Chen YP, Chan ATC, Le QT, Blanchard P, Sun Y, Ma J: Nasopharyngeal carcinoma. Lancet 2019, 394(10192): 64-80.
- Sakai H, Sato A, Aihara Y, Ikarashi Y, Midorikawa Y, Kracht M, Nakagama H, Okamoto K: MKK7 mediates miR-493-dependent suppression of liver metastasis of colon cancer cells. Cancer Sci 2014, 105(4): 425-430.
- Lacombe ML, Milon L, Munier A, Mehus JG, Lambeth DO: The human Nm23/nucleoside diphosphate kinases. J Bioenerg Biomembr 2000, 32(3): 247-258.
- Singh LS, Berk M, Oates R, Zhao Z, Tan H, Jiang Y, Zhou A, Kirmani K, Steinmetz R, Lindner D, et al: Ovarian cancer G protein-coupled receptor 1, a new metastasis suppressor gene in prostate cancer. J Natl Cancer Inst 2007, 99(17): 1313-1327.
- Wang X, Wang S, Tang X, Zhang A, Grabinski T, Guo Z, Hudson E, Berghuis B, Webb C, Zhao P, et al: Development and evaluation of monoclonal antibodies against phosphatidylethanolamine binding protein 1 in pancreatic cancer patients. J Immunol Methods 2010, 362(1-2): 151-160.
- Griner EM, Theodorescu D: The faces and friends of RhoGDI2. Cancer Metastasis Rev 2012, 31(3-4): 519-528.
- Eves EM, Shapiro P, Naik K, Klein UR, Trakul N, Rosner MR: Raf kinase inhibitory protein regulates aurora B kinase and the spindle checkpoint. Mol Cell 2006, 23(4): 561-574.
- Gautam A, Li ZR, Bepler G: RRM1-induced metastasis suppression through PTEN-regulated pathways. Oncogene 2003, 22(14): 2135-2142.
- Javelaud D, Mohammad KS, McKenna CR, Fournier P, Luciani F, Niewolna M, André J, Delmas V, Larue L, Guise TA, et al: Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res 2007, 67(5): 2317-2324.
- Xia W, Unger P, Miller L, Nelson J, Gelman IH: The Src-suppressed C kinase substrate, SSeCKS, is a potential metastasis inhibitor in prostate cancer. Cancer Res 2001, 61(14): 5644-5651.
- Morrison JA, Pike LA, Sams SB, Sharma V, Zhou Q, Severson JJ, Tan AC, Wood WM, Haugen BR: Thioredoxin interacting protein (TXNIP) is a novel tumor suppressor in thyroid cancer. Mol Cancer 2014, 19(13): 62-79.
- Blume-Jensen P, Hunter T: Oncogenic kinase signalling. Nature 2001, 411(6835): 355-365.
- Crossman SH, Janovjak H: Light-activated receptor tyrosine kinases: Designs and applications. Curr Opin Pharmacol 2022, 63(5): 102197-102208.
- Black PC, Dinney CP: Bladder cancer angiogenesis and metastasis--translation from murine model to clinical trial. Cancer Metastasis Rev 2007, 26(3-4): 623-634.
- Dinney CP, Fishbeck R, Singh RK, Eve B, Pathak S, Brown N, Xie B, Fan D, Bucana CD, Fidler IJ, et al: Isolation and characterization of metastatic variants from human transitional cell carcinoma passaged by orthotopic implantation in athymic nude mice. J Urol 1995, 154(4): 1532-1538.
- Mooso BA, Vinall RL, Mudryj M, Yap SA, deVere White RW, Ghosh PM: The role of EGFR family inhibitors in muscle invasive bladder cancer: a review of clinical data and molecular evidence. J Urol 2015, 193(1): 19-29.
- Fleischmann A, Rotzer D, Seiler R, Studer UE, Thalmann GN: Her2 amplification is significantly more frequent in lymph node metastases from urothelial bladder cancer than in the primary tumours. Eur Urol 2011, 60(2): 350-357.
- McConkey DJ, Choi W, Marquis L, Martin F, Williams MB, Shah J, Svatek R, Das A, Adam L, Kamat A, et al: Role of epithelial-to-mesenchymal transition (EMT) in drug sensitivity and metastasis in bladder cancer. Cancer Metastasis Rev 2009, 28(3-4): 335-344.
- He L, Kang Q, Chan KI, Zhang Y, Zhong Z, Tan W: The immunomodulatory role of matrix metalloproteinases in colitis-associated cancer. Front Immunol 2023, 13(19): 1093990.
- Folkman J, Watson K, Ingber D, Hanahan D: Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature 1989, 339(6219): 58-61.
- Blood CH, Zetter BR: Tumor interactions with the vasculature: angiogenesis and tumor metastasis. Biochim Biophys Acta 1990, 1032(1): 89-118.
- Karelina TV, Goldberg GI, Eisen AZ: Matrix metalloproteinases in blood vessel development in human fetal skin and in cutaneous tumors. J Invest Dermatol 1995, 105(3): 411-417.
- Chaffer CL, Dopheide B, McCulloch DR, Lee AB, Moseley JM, Thompson EW, Williams ED: Upregulated MT1-MMP/TIMP-2 axis in the TSU-Pr1-B1/B2 model of metastatic progression in transitional cell carcinoma of the bladder. Clin Exp Metastasis 2005, 22(2): 115-125.
- Acuff HB, Carter KJ, Fingleton B, Gorden DL, Matrisian LM: Matrix metalloproteinase-9 from bone marrow-derived cells contributes to survival but not growth of tumor cells in the lung microenvironment. Cancer Res 2006, 66(1): 259-266.
- Jeon S, Kim TK, Jeong SJ, Jung IH, Kim N, Lee MN, Sonn SK, Seo S, Jin J, Kweon HY, et al: Anti-Inflammatory Actions of Soluble Ninjurin-1 Ameliorate Atherosclerosis. Circulation 2020, 142(18): 1736-1751.
- Huang S, Van Arsdall M, Tedjarati S, McCarty M, Wu W, Langley R, Fidler IJ: Contributions of stromal metalloproteinase-9 to angiogenesis and growth of human ovarian carcinoma in mice. J Natl Cancer Inst 2002, 94(15): 1134-1142.
- Itoh T, Tanioka M, Yoshida H, Yoshioka T, Nishimoto H, Itohara S: Reduced angiogenesis and tumor progression in gelatinase A-deficient mice. Cancer Res 1998, 58(5): 1048-1051.
- Orgaz JL, Herraiz C, Sanz-Moreno V: Rho GTPases modulate malignant transformation of tumor cells. Small GTPases 2014, 5(2): e29019.
- Jaffe AB, Hall A: Rho GTPases in transformation and metastasis. Adv Cancer Res 2002, 84(51): 57-80.
- Cooke M, Baker MJ, Kazanietz MG, Casado-Medrano V: PKC regulates Rho GTPases and actin cytoskeleton reorganization in non-small cell lung cancer cells. Small GTPases 2021, 12(3): 202-208.
- Hanna S, El-Sibai M: Signaling networks of Rho GTPases in cell motility. Cell Signal 2013, 25(10): 1955-1961.
- Nobes CD, Hall A: Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81(1): 53-62.
- Cai X, Liu Y, Yang W, Xia Y, Yang C, Yang S, Liu X: Long noncoding RNA MALAT1 as a potential therapeutic target in osteosarcoma. J Orthop Res 2016, 34(6): 932-941.
- Zhang F, Li J, Xiao H, Zou Y, Liu Y, Huang W: AFAP1-AS1: A novel oncogenic long non-coding RNA in human cancers. Cell Prolif 2018, 51(1): e12397.
- Gildea JJ, Seraj MJ, Oxford G, Harding MA, Hampton GM, Moskaluk CA, Frierson HF, Conaway MR, Theodorescu D: RhoGDI2 is an invasion and metastasis suppressor gene in human cancer. Cancer Res 2002, 62(22): 6418-6423.
- Huang H, Jin H, Zhao H, Wang J, Li X, Yan H, Wang S, Guo X, Xue L, Li J, et al: RhoGDI promotes Sp1/MMP-2 expression and bladder cancer invasion through perturbing miR-200c-targeted JNK2 protein translation. Mol Oncol 2017, 11(11): 1579-1594.
- Greenburg G, Hay ED: Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J Cell Biol 1982, 95(1): 333-339.
- Czerniak B, Dinney C, McConkey D: Origins of Bladder Cancer. Annu Rev Pathol 2016, 23(11): 149-174.
- Banyard J, Bielenberg DR: The role of EMT and MET in cancer dissemination. Connect Tissue Res 2015, 56(5): 403-413.
- Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL, et al: Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145(6): 926-940.
- Cheung KJ, Gabrielson E, Werb Z, Ewald AJ: Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013, 155(7): 1639-1651.
- Labelle M, Begum S, Hynes RO: Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20(5): 576-590.
- Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW, Williams ED: Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res 2006, 66(23): 11271-11278.
- Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J: Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 2012, 22(6): 725-736.
- Franzen CA, Blackwell RH, Todorovic V, Greco KA, Foreman KE, Flanigan RC, Kuo PC, Gupta GN: Urothelial cells undergo epithelial-to-mesenchymal transition after exposure to muscle invasive bladder cancer exosomes. Oncogenesis 2015, 4(8): e163.
- Rao SS, Kondapaneni RV, Narkhede AA: Bioengineered models to study tumor dormancy. J Biol Eng 2019, 10(13): 3-15.
- Hensel JA, Flaig TW, Theodorescu D: Clinical opportunities and challenges in targeting tumour dormancy. Nat Rev Clin Oncol 2013, 10(1): 41-51.
- Horak CE, Lee JH, Marshall JC, Shreeve SM, Steeg PS: The role of metastasis suppressor genes in metastatic dormancy. APMIS 2008, 116(7-8): 586-601.
- Horak CE, Lee JH, Marshall JC, Shreeve SM, Steeg PS: The role of metastasis suppressor genes in metastatic dormancy. APMIS 2008, 116(7-8): 586-601.
- Titus B, Frierson HF Jr, Conaway M, Ching K, Guise T, Chirgwin J, Hampton G, Theodorescu D: Endothelin axis is a target of the lung metastasis suppressor gene RhoGDI2. Cancer Res 2005, 65(16): 7320-7327.
Annals of urologic oncology
p-ISSN: 2617-7765, e-ISSN: 2617-7773
 Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.