
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
Hereditary Renal Cell Tumors: Clinicopathologic Importance
Harmanjot Singh1, Mukul K. Divatia1, Donghwa Baek1, Jae Y. Ro1
1Department of Pathology and Genomic Medicine, Houston Methodist Hospital, Weill Medical College of Cornell University, Houston, Texas, 77030, USA. Correspondence: Jae Y. Ro (Department of Pathology and Genomic Medicine, Houston Methodist Hospital, 6565 Fannin Street, Houston, Texas, 77030 USA; Email: jaero@houstonmethodist.org).
Annals of Urologic Oncology 2021, 4(1): 18-30. https://doi.org/10.32948/auo.2021.10.15
Received: 30 Jun 2021 | Accepted: 07 Oct 2021 | Published online: 15 Oct 2021 
Hereditary renal cancer syndromes represent approximately 5% of renal malignancies and have distinctive clinical, histopathologic, and genetic features. Next-generation sequencing and other molecular testing methods have uncovered several hereditary renal cancer syndromes. Several autosomal dominant hereditary renal cell carcinoma (RCC) syndromes, including those related to germline pathogenic variants in VHL, BAP1, MITF, MET, FH, TSC1/TSC2, FLCN, SDH, and CDC73 have been confirmed. FH- and BAP1-related RCCs are associated with more aggressive disease. Identifying the clinical and pathological features in these hereditary RCC syndromes is important as, relative to familial cohorts, these patients require early screening and intervention and regular surveillance to improve their clinical prognosis and long-term outcomes. More importantly, identification of these syndromes plays a vital role in personalized management and systemic treatment selection in this modern era of precision medicine. Ongoing studies have demonstrated that treatment based on genetic pathway targeting is a promising approach for hereditary renal cancer management. This review describes updates in the diagnostic criteria for and management of familial kidney cancer syndromes.
Key words hereditary cancer, kidney cancer, renal cancer syndrome, renal cell carcinoma, von Hippel-Lindau disease, tuberous sclerosis complex, hereditary leiomyomatosis-associated renal cell carcinoma
Von Hippel-Lindau (VHL), hereditary papillary RCC (pRCC), and Birt-Hogg-Dube (BHD) syndromes are the most frequently encountered entities presenting with bilateral, multifocal tumors. Hereditary leiomyomatosis, RCC syndrome-associated RCC (HLRCC), and succinate dehydrogenase (SDH)-deficient RCCs typically present with unilateral and solitary tumor. Less common hereditary renal cancer syndromes include tuberous sclerosis complex-associated RCCs, BAP1 tumor predisposition syndrome, constitutional chromosome 3 translocations, PTEN hamartoma/Cowden syndrome, and hyperparathyroidism-jaw tumor syndrome. Major advancements in our understanding of the molecular pathogenesis of RCC, including the identification of several new mutations, have provided a means to offer targeted treatment options and improve overall patient management. Through genetic testing, at-risk individuals can be identified earlier in their disease course and offered regular surveillance, potentially improving prognosis and outcomes. In this review, we provide a comprehensive outline of the genetic and clinical features of well-recognized and emerging hereditary RCC syndromes.
VHL syndrome is an inherited autosomal dominant disorder caused by constitutional mutations in the VHL tumor suppressor gene due to either a missense mutation or deletion in the short arm of chromosome 3. VHL has an incidence of 1 in 30,000 [2, 3] with more than 90% of patients developing manifestations by age 65 [2].
The VHL gene product regulates the hypoxic gene response by stabilizing alpha subunits of hypoxia-inducible factor (HIF)-1 and HIF-2 transcription factors [4-6]. Mutated VHL protein does not bind to HIF-1α and 2α, resulting in gene transcription and upregulation of growth factors, including erythropoietin, vascular endothelial growth factor (VEGF), platelet-derived growth factor β, and other genes involved in metabolism. Thus, VHL mutation drives angiogenesis and proliferation.
VHL mutations result in an increased propensity to develop a multitude of benign and malignant tumors, including hemangioblastomas of the retina and central nervous system, and clear cell RCC (ccRCC) (over 70% each) [2, 7]. Less frequently seen tumors include pheochromocytoma/paraganglioma (20%), pancreatic neuroendocrine tumor (10%), and endolymphatic sac tumor (5-10%). Additionally, multiple visceral cysts, including renal, pancreatic, epididymal, and parametrial cysts are commonly seen.
VHL may demonstrate phenotypic variation. Patients with truncating mutations (or type 1 mutations) have a lower incidence of pheochromocytoma compared to patients with missense mutations (or type 2 mutations) [7-10]. VHL frameshift and nonsense mutations are associated with high penetrance of ccRCC, whereas full or partial deletions confer a lower risk [7]. Genetic testing for VHL mutations detects nearly 100% of individuals with VHL [11]. VHL mutations are found in both inherited and sporadic ccRCCs, with evidence of both copies lost in 86% of cases [12]. Apart from the shared feature of 3p loss, tumors within the same patient are clonally diverse, highlighting the development of different molecular pathways that initiate further tumorigenesis following the loss of 3p. Studies have identified two subgroups of VHL-inactivated clear cell malignancies: those with a HIF-1α and 2α-driven genotype and those with a HIF-2α-dominant genotype [13, 14]. The discovery of these genotypes and their implications for particular metabolic pathways has helped drive the development of novel therapies. Additionally, familial genetic screening with routine imaging can lower morbidity and mortality. As a result of increased awareness and increasingly aggressive approaches, approximately 85-90% of VHL patients are diagnosed with renal masses sized less than 6 cm, with only 11% developing distant metastases [15].
Renal lesions in VHL are commonly bilateral and multicentric. Grossly, the kidney often shows multiple small cystic and solid lesions, with the number of cysts ranging from 3 to 30 (on average, 7.8 cysts per kidney) with an average size of 0.7 cm [16].
Histologic features of renal tumors in VHL syndrome include simple cysts lined by one to two layers of clear cells with bland features, atypical cysts with more complex epithelial proliferations, and scattered aggregates of clear cell proliferation in renal parenchyma to ccRCC arising either within cysts or even from non-cystic kidney parenchyma. These tumors tend to be low-grade and show identical morphological and immunohistochemical features to sporadic ccRCCs, characterized by nests of epithelial cells with abundant clear cytoplasm with a delicate sinusoidal vascular network (Figure 1). In addition, the adjacent renal parenchyma often shows microscopic foci of clear cell clusters or proliferations with unilocular cysts lined by clear cells without significant cytologic atypia. However, cysts lined by multiple layers of clear cells or focal papillary proliferation with or without cytologic atypia may also be seen [17].
Tumors with typical features of clear cell papillary RCC (ccpRCC)—papillary architecture harboring nuclei with a linear arrangement along the apical side of the tumor cells away from the basement membrane—can be seen in VHL disease [18, 19]. However, tumors with ccpRCC-like morphology are molecularly distinct from sporadic ccpRCC. In addition to an established family history and/or strong clinical suspicion, the combination of bilateral, multifocal ccpRCCs, clusters of clear cell proliferations in adjacent renal parenchyma, and multiple predominantly clear cell-lined cysts are helpful pathologic features to distinguish VHL syndrome from sporadic cases of ccpRCC. These tumors rarely metastasize, and small tumors are typically managed conservatively, while larger tumors (>3 cm) are excised or ablated.
|
Table 1. Overview of Hereditary Renal Cell Cancer Syndromes. |
||||||
|
Syndrome |
Inheritance |
Gene |
Estimated risk of developing RCC |
Histological subtype of renal tumors |
Effect of mutation |
Extrarenal features |
|
VHL disease |
AD |
VHL |
70% |
ccRCC ccpRCC |
? Increased angiogenesis and proliferation through activation of hypoxia response pathways |
? CNS and retinal hemangioblastomas ? Endolymphatic sac tumor ? Pancreatic cysts and neuroendocrine tumors ? Pheochromocytomas |
|
Hereditary pRCC |
AD |
MET |
Increased |
pRCC type 2 |
? MET signaling pathway activation |
? None known |
|
HLRCC |
AD |
FH |
15-20% |
pRCC type 2 Collecting duct RCC tcRCC |
? Activation of hypoxia response pathways ? Epigenetic effects |
? Uterine and cutaneous leiomyomas ? Leiomyosarcomas |
|
BHD syndrome |
AD |
FLCN |
25-35% |
HOCT ccRCC pRCC Multifocal oncocytosis |
? mTOR pathway activation ? Increased mitochondrial biogenesis and replication ? Dysregulation of cell-cell adhesion |
? Cutaneous fibrofolliculomas and other skin adnexal tumors |
|
TSC |
AD |
TSC1 TSC2 |
2-4% |
Angiomyolipoma ccRCC HOCT |
? Upregulation and activation of mTOR pathway |
? Cortical dysplasia ? Rhabdomyomas ? Subependymal giant cell astrocytoma ? Angiomyofibromas and cutaneous lesions ? Lymphangioleiomyomatosis ? Neural tumors |
|
SDH-deficient RCC |
AD |
SDHA SDHB SDHC SDHD |
10-15% |
ccRCC chRCC Oncocytoma |
? Accumulation of succinate with subsequent activation of hypoxia response |
? Pheochromocytoma ? Paraganglioma ? GIST |
|
HPT-JT syndrome |
AD |
HRPT2 |
15% |
Nephroblastoma (adult Wilms tumor) Mixed epithelial and stromal tumor pRCC Hamartoma |
? Disruption of chromatin remodeling ? Activation of Wnt signaling pathway |
? Primary hyperparathyroidism ? Parathyroid carcinoma ? Ossifying fibromas of maxilla and mandible ? Uterine tumors |
|
Constitutional chromosome 3 translocations |
Chromosomal |
|
Up to 70% |
ccRCC |
? Loss of chromosome 3p resulting in activation of hypoxia response pathways |
? None |
|
BAP1 tumor predisposition |
AD |
BAP1 |
10-12% |
ccRCC |
? Disruption in chromatin remodeling and cell cycle regulation |
? Cutaneous and uveal melanomas ? Malignant mesothelioma ? Cholangiocarcinoma |
|
PTEN hamartoma/ Cowden syndrome |
AD |
PTEN |
Up to 35% |
pRCC chRCC ccRCC |
? Activation and upregulation of PI3K signaling pathways |
? Thyroid cancer ? Breast cancer ? Endometrial cancer ? Colon cancer ? Cutaneous and mucosal hamartomas |
|
AD, autosomal dominant; BHD, Birt-Hogg-Dubé; ccpRCC, clear cell papillary renal cell carcinoma; ccRCC, clear cell renal cell carcinoma; chRCC, chromophobe renal cell carcinoma; HLRCC, hereditary leiomyomatosis-associated renal cell carcinoma; HPT-JT, hyperparathyroidism-jaw tumor; GIST, gastrointestinal stromal tumor; HOCT, hybrid oncocytic/chromophobe renal cell carcinoma; PI3K, phosphoinositide 3-kinase; pRCC, papillary renal cell carcinoma; RCC, renal cell carcinoma; tcRCC, tubulocystic renal cell carcinoma; SDH, succinate dehydrogenase; TSC, tuberous sclerosis complex; VHL, von Hippel-Lindau |
||||||
|
Table 2. Screening Recommendations for Hereditary Renal Cell Carcinoma syndromes. |
||
|
Syndrome |
Screening Recommendations- Renal |
Screening Recommendations- Systemic |
|
VHL disease |
Abdominal MRI imaging every 1-2 years [95] |
· Ophthalmologic assessment · Blood pressure check · Neurologic examination · Audiology examination · Urinary metanephrines /Plasma free metanephrines · CNS and spinal MRI imaging |
|
Hereditary pRCC |
Abdominal CT/MRI imaging every 2 years |
· None |
|
HLRCC |
Annual MRI imaging with kidney protocol [96] |
· Dermatologic check-up · Gynecological examination with radiologic studies for females |
|
BHD syndrome |
Abdominal CT/MRI imaging every 3 years [97] |
· Dermatologic check-up · CT scan of thorax |
|
TSC |
Abdominal CT/MRI imaging every 1-3 years [98] |
· Dermatologic check-up, annual · Dental exam, six months · ECG · Ophthalmological assessment · Annual blood pressure check · Annual kidney function tests · Neuropsychiatric disorder screening · Brain MRI · EEG, if clinically indicated · CT thorax, every 5–10 years |
|
SDH-deficient RCC |
Abdominal CT/MRI imaging every 2 years
|
· Blood pressure check · Plasma methoxytyramine and free metanephrines /urine metanephrines · Serum chromogranin A · Complete blood counts · Whole-body MRI · Neck MRI, if clinically indicated |
|
HPT-JT syndrome |
Abdominal CT/MRI imaging every 5 years [99] |
· Serum calcium, intact parathyroid hormone, and 25-(OH) vitamin D levels · Dental panoramic X-ray · Radiologic screening for uterine tumors in females |
|
Constitutional chromosome 3 translocations |
Abdominal CT/MRI imaging every 1-3 years |
· None |
|
BAP1 tumor predisposition |
Abdominal CT/MRI imaging every 2 years [100] |
· Dermatologic check-up · Ophthalmologic assessment · CT or MRI of thorax/abdomen, if clinically indicated |
|
PTEN hamartoma/ Cowden syndrome |
Abdominal CT/MRI imaging every 1-2 years [101] |
· Dermatologic check-up · Thyroid assessment with ultrasound · Breast exam, mammogram and breast MRI in females · Colonoscopy · Endometrial cancer screening in females |
|
BHD, Birt-Hogg-Dubé; CT, computed tomography; ECG, electrocardiogram; EEG, electroencephalogram; HLRCC, hereditary leiomyomatosis-associated renal cell carcinoma; HPT-JT, hyperparathyroidism-jaw tumor; MRI, magnetic resonance imaging; RCC, renal cell carcinoma; SDH, succinate dehydrogenase; TSC, tuberous sclerosis complex; VHL, von Hippel-Lindau |
||
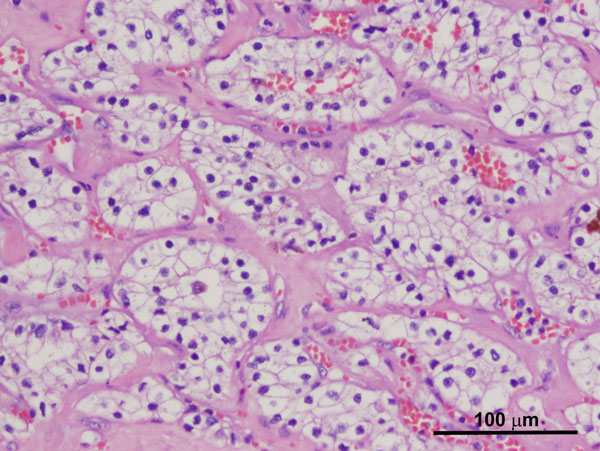
Figure 1. Conventional ccRCC with characteristic cytoarchitectural features associated with VHL syndrome (H and E, 100x).
Hereditary pRCC is an extremely rare autosomal dominantly inherited disorder due to activating mutations in the MET proto-oncogene on chromosome 7p31 [20-22] with incomplete penetrance. The incidence is 1 in 10 million [23]. The MET gene encodes a receptor tyrosine kinase involved in cell signaling and growth. These mutations predispose individuals to increased risk for multiple, bilateral pRCC type 1 [20]. Peculiarly, these tumors tend to manifest at a later age (50-70 years) with high penetrance but may also be seen in patients as young as 30 years. In contrast to other hereditary RCC syndromes, there are no known extrarenal manifestations.
On gross examination, numerous well-circumscribed bilateral and multifocal tumors are present. Morphologically, these tumors demonstrate a papillary or tubulopapillary architecture, with papillae lined by cuboidal epithelial cells with small to scant cytoplasm, small oval to round low-grade nuclei, and inconspicuous nucleoli. Most tumors are WHO/ISUP grade 1 or 2, although some grade 3 tumors may also be seen. Foamy macrophages and psammomatous calcifications are commonly noted. Clear cells are not uncommon and are most prominent in tumors with necrosis and hemorrhage. Rarely, focal eosinophilic cytoplasmic change can be seen mimicking the morphology seen in type 2 pRCC. The adjacent non-neoplastic renal parenchyma often shows numerous papillary adenomas. In contrast to patients with sporadic pRCC, patients with hereditary pRCC often show dozens to hundreds of lesions in the kidney. Additionally, the presence of numerous papillary adenomas in the adjacent parenchyma helps distinguish hereditary pRCC from sporadic.
Hereditary leiomyomatosis and RCC syndrome-associated RCC (HLRCC) show an autosomal dominant pattern of inheritance with an incidence of approximately 1 in 200,000 [24]. HLRCC is caused by a germline mutation in the fumarate hydratase (FH) gene located on chromosome 1q42. FH encodes an enzyme responsible for catalyzing the conversion of fumarate to malate in the Krebs cycle [25-28]. This mutation subsequently leads to an accumulation of intracellular fumarate, which mediates various proteomic and epigenetic events, ultimately resulting in activation of HIF-1. Approximately 71-100% of families with features suggestive of HLRCC demonstrate pathogenic variants [25, 26, 29].
Extrarenal manifestations include multiple cutaneous leiomyomas and early onset uterine leiomyomas and leiomyosarcomas. Uterine leiomyomas are seen in 70-80% of females with HLRCC [28]. Clinically, these patients may present with a history of menorrhagia and pelvic pressure or pain and frequently require hysterectomy before 30 years of age [31]. Cutaneous leiomyomas often present in the second to fourth decade of life, presenting as painful red-brown papules or nodules on the trunk or extremities. These cutaneous lesions rarely undergo malignant transformation [32, 33]. Renal malignancies present in approximately 15-20% of patients with HLRCC with early onset (36-39 years) [32, 34, 35]. Lower stage tumors in HLRCC may metastasize. HLRCC-associated RCC is usually aggressive and frequently presents with metastatic disease involving lymph nodes, bone, brain, lungs, or other body sites.
HLRCC syndrome-associated RCC has been included as a new tumor entity in the most recent WHO classification of kidney tumors. On gross examination, the renal tumors seen in HLRCC are often unilateral and solitary with features varying from solid and homogenous to cystic with areas of hemorrhage and necrosis. The adjacent non-neoplastic renal parenchyma is often unremarkable or demonstrates few cysts lined by eosinophilic to clear epithelium.
These tumors often exhibit several growth patterns, including papillary, tubulopapillary, tubular, cribriform, and tubulocystic architectures. High-grade or de-differentiated foci, solid and cystic components, sarcomatoid differentiation, and collecting duct carcinoma-like areas can also be seen. A characteristic cytological feature of HLRCC-associated RCCs is a notably prominent eosinophilic nucleolus surrounded by a clear halo, though this finding is not entirely specific or sensitive for establishing this particular diagnosis (Figure 2A) [30, 36]. HLRCC-associated RCCs are often of high nuclear grade (WHO/ISUP grade 3-4) [34, 36]. Immunohistochemistry shows loss of FH within tumor cells (Figure 2B) in the majority of cases while an immunostain for 2-succinyl-cysteine (2SC), an oncometabolite derived from fumarate, shows diffuse nuclear and cytoplasmic staining, which is highly correlated with the presence of an FH mutation.
HLRCC is associated with a poor prognosis with a major subset of cases presenting with metastatic disease. Therefore, accurate recognition and early diagnosis of HLRCC is critical for effective management. Definitive confirmation can be obtained by testing for a germline FH mutation with genetic counseling for family members. Renal cancer surveillance is recommended at an early age for familial cases.
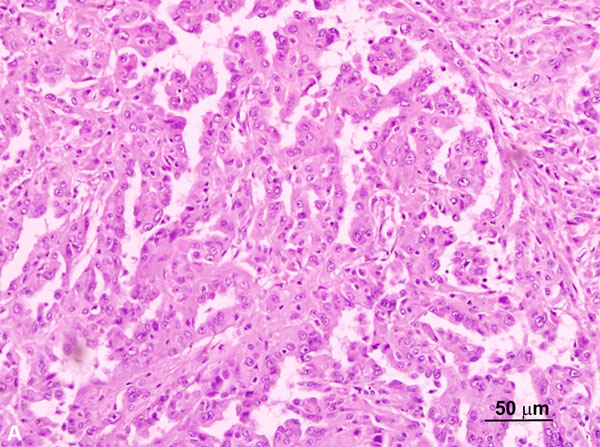
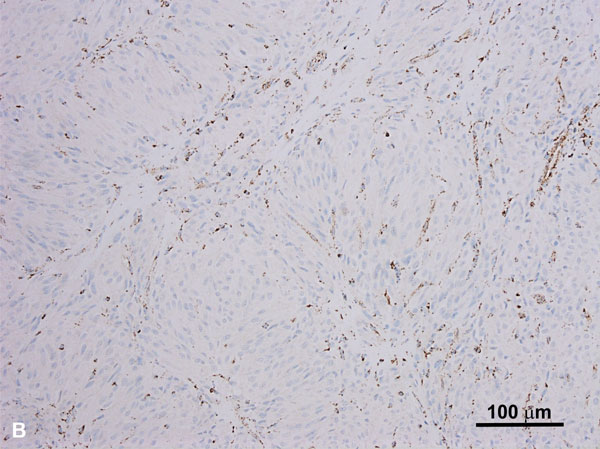
Figure 2A. HLRCC-associated RCC case with a papillary growth pattern (H and E, 200x). Note the tumor cells with prominent enlarged nucleoli. Figure 2B. HLRCC-associated RCC with loss of FH in tumor cells (Immunoperoxidase, 100x).The stromal cells serve as an internal control and show no loss of FH expression.
BHD syndrome is a rare autosomal dominantly inherited disorder caused by point mutations and rearrangements of the folliculin (FLCN) gene located on chromosome 17p11.2 [37, 38]. The function of FLCN is controversial but it has been suggested that it functions as a tumor suppressor and is involved in cell-cell adhesion, negative regulation of ribosomal RNA synthesis, and mTOR pathway modulation [39-42]. BHD syndrome is estimated to be seen in approximately 1 in 2 million population [43].
BHD is characterized by the development of multiple kidney tumors, facial fibrofolliculomas, and pulmonary cysts [44, 45]. Along with having an increased risk of developing kidney tumors, which are observed in 25-35% of patients [46-49], these patients also have a significantly higher risk of developing a spontaneous pneumothorax [50]. A wide variety of renal malignancies have been identified in BHD. Hybrid oncocytic tumors, characterized as being mixed oncocytoma and chromophobe RCCs (chRCC), are the most common (Figure 3) [50]. Patients diagnosed with hybrid oncocytic tumors should be evaluated for BHD, as these are characteristic of this disease. Other renal tumors include clear cell RCC, pRCC, angiomyolipoma, and oncocytoma (including multifocal oncocytosis) [51]. Major criteria for BHD diagnosis include: (1) at least five fibrofolliculomas, at least one histologically confirmed, of adult onset, or (2) identified pathogenic FLCN mutation. Minor criteria include: (1) multiple pulmonary cysts with no other apparent cause, with or without spontaneous pneumothorax, (2) early onset (<50 years) of renal malignancy that is multifocal, bilateral, or mixed histology (chromophobe and oncocytic histology), and (3) a first-degree relative with BHD [52]. For BHD diagnosis, patients should have one major or two minor criteria present.
On gross examination, 50% of patients present with bilateral and multifocal tumors with a homogeneous tan-brown cut surface without hemorrhage or necrosis, with or without a central scar [53]. These tumors are typically small at presentation with low metastatic potential. Morphologically, hybrid oncocytic tumors are most commonly encountered with areas resembling chRCC or oncocytoma, or tumor cells showing irregular nuclear contours without perinuclear halos. Oncocytic tumors in BHD syndrome can show nests of clear cells in an otherwise typical oncocytic tumor. Tumors histologically resembling chRCC, ccRCC, pRCC, and renal oncocytoma have been described. Evaluation of the surrounding renal parenchyma for features of oncocytosis, including numerous microscopic oncocytic nodules, cysts lined by oncocytic cells, or oncocytic cells between native renal parenchyma is essential. The characteristic nuclear feature of sporadic chRCCs—hyperchromatic, raisinoid nuclei with perinuclear halos—is less often seen or only focally present in the hybrid oncocytic tumors of BHD.
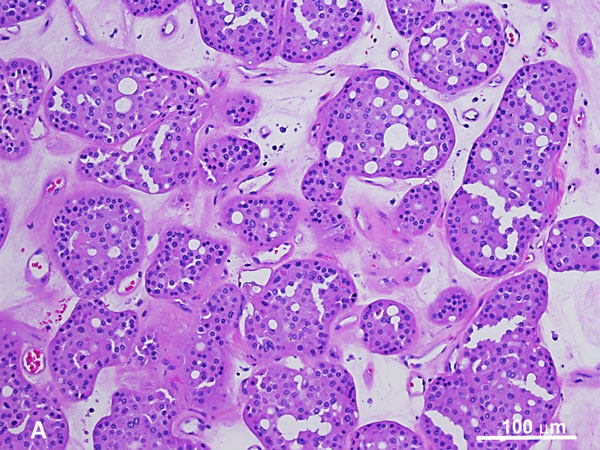
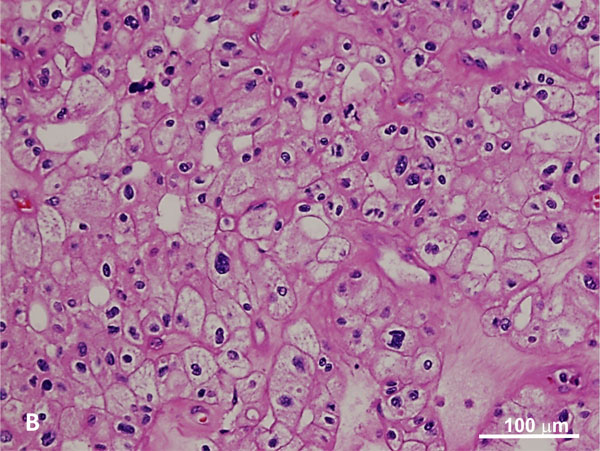
Figure 3. Hybrid tumor with features of oncocytoma (A) and chromophobe RCC (B) in BHD syndrome. The oncocytic tumor exhibits a nested growth pattern with round low-grade nuclei whereas the chromophobe RCC component shows distinct cell membrane, characteristic perinuclear haloes with binucleation and raisinoid nuclei (H and E, 100x).
Tuberous sclerosis complex (TSC) is an autosomal dominantly inherited disorder characterized by formation of hamartomas in numerous organs including brain, skin, kidney, and lung. TSC patients exhibit associated cutaneous involvement, including hypopigmented macules, facial angiofibromas, and periungual fibromas in almost all cases. Most affected patients also show features of central nervous system involvement, including cortical dysplasia, subependymal nodules, and, less commonly, subependymal giant cell astrocytomas with other neurological conditions, including seizures. Other manifestations include renal angiomyolipoma (AML), lymphangioleiomyomatosis, cardiac rhabdomyomas, and retinal hamartomas.
TSC is caused by inactivating mutations of the TSC1 gene located on chromosome 9q34 and the TSC2 gene located on chromosome 16p13.3. TSC1 and TSC2 encode hamartin and tuberin, respectively. These proteins are responsible for inhibiting downstream pathways of mammalian target of rapamycin (mTOR) [54]. Inactivation of either of these proteins results in upregulation of the HIF pathway. In 70% of cases, these mutations occur as spontaneous germline mutations [55]. Patients with TSC2 mutations appear to be more severely affected and show greater kidney involvement.
Renal manifestations of TSC include multiple, bilateral, AMLs (75-80%), oncocytomas, and rarely RCC (2-4%) [56, 57]. Grossly, multiple simple cysts and tan-yellow nodules representing AMLs are frequently seen. In approximately 5% of patients, diffuse cystic disease similar to that seen in autosomal dominant polycystic disease can develop. Morphologically, TSC-associated RCCs are multifocal with multiple AMLs and cysts, and a wide spectrum of architectural patterns. Some tumors show clear and voluminous cytoplasm with papillary, acinar, tubular, and alveolar architecture resembling ccRCC and ELOC/TCEB1-mutated RCCs with prominent fibromuscular stroma. Other tumors show nests or sheets of eosinophilic or oncocytic tumor cells resembling chRCC, oncocytoma, or hybrid oncocytic tumors. The remaining TSC-associated tumors often show unclassifiable features characterized by eosinophilic tumor cells resembling eosinophilic solid and cystic RCC, sometimes with high-grade nuclear features. Most tumors show a mixed architectural pattern. Sporadic RCCs characterized by somatic TSC1/TSC2/MTOR mutations demonstrating significant morphological overlap with tumors in TSC cases have been reported recently. TSC-associated RCCs generally are clinically indolent with rare metastasis and these tumors are not typically associated with increased mortality.
The succinate dehydrogenase (SDH) complex, a component of the Krebs cycle and the electron transport chain, helps convert succinate to fumarate. Dysfunctions in SDH result in elevated levels of succinate and subsequent inhibition of HIF-α prolyl hydroxylases leading to the induction of hypoxia response pathways. Germline mutations in genes encoding SDH protein products, including SDHA/B/C/D are associated with an increased risk of cancer development and responsible for familial paraganglioma-pheochromocytoma syndromes, gastrointestinal stromal tumors (GIST), pituitary adenomas, and RCCs. SDHB is most frequently involved in SDH-deficient RCC [58, 59]. The incidence of RCC is approximately 0.05% to 0.1% of all renal tumors with a mean age of presentation of 40 years [58]. These tumors more frequently demonstrate early metastasis and appear to be more aggressive compared to other hereditary renal tumors. The most commonly reported renal tumors in these patients include ccRCC, chRCC, and oncocytomas [60, 61]. SDH-deficient RCC has also been added as a new tumor entity in the WHO 2016 classification of kidney tumors. These tumors are characterized by a loss of SDHB immunoreactivity and patients with SDH-deficient RCC often have a mutation in SDHB, although other SDH mutations have been reported.
Grossly, SDHB-deficient RCCs are often solitary and unilateral, with bilateral tumors observed in 26% of reported cases [23]. They are well-circumscribed with a pushing border and may be associated with a pseudocapsule. Rarely, they may show an infiltrative border. Upon gross examination, they often appear solid with a red-brown cut surface. Morphologically, they show predominantly solid and cystic architecture with vacuolated cytoplasm and cytoplasmic inclusions containing pale eosinophilic flocculent material, along with cytoplasmic inclusions (Figure 4A) that can be identified as giant mitochondria upon ultrastructural examination. In conjunction with SDH immunostaining, histologic features, such as monomorphic oncocytic renal tumors with a solid growth pattern, intratumoral mast cells, and cytoplasmic inclusions with flocculent material, can be used to support diagnosis. High-grade features including sarcomatoid differentiation, high nuclear grade, and necrosis are uncommon.
Immunohistochemical studies show a loss of SDHB reactivity (Figure 4B), although a lack of SDHB staining can be seen in tumors with mutations in SDHA, SDHC, SDHD, or SDHAF2. In addition, these tumors are often negative for CK7 and CD117 (c-kit). Many of these tumors show indolent clinical behavior, but up to 33% can show metastasis and an aggressive clinical course [23]. Cases that do show metastasis or an aggressive clinical course are often associated with high-grade features.
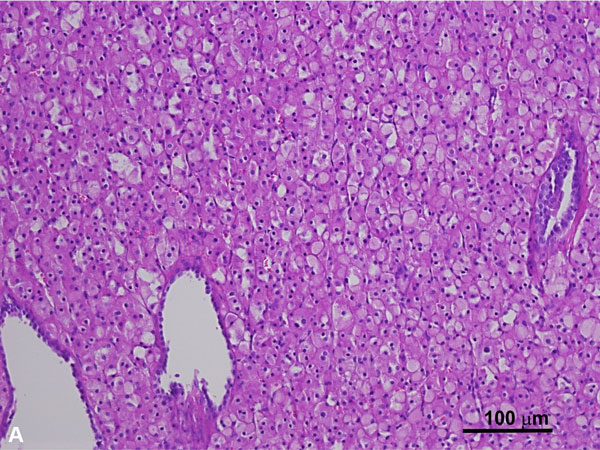
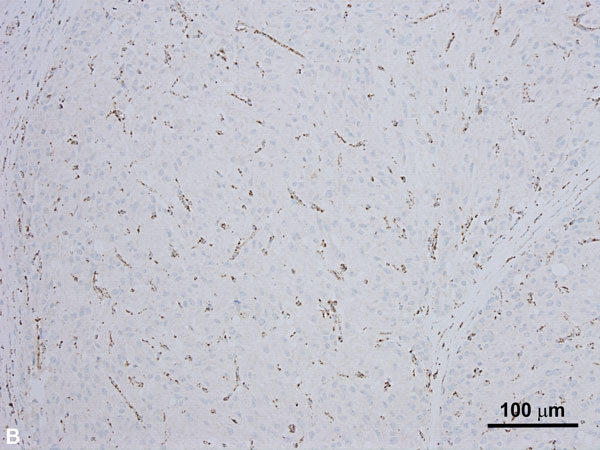
Figure 4A. SDH-deficient RCC demonstrating cells with uniform nuclei, fine stippled chromatin and flocculent eosinophilic cytoplasm (H and E, 100x). Figure 4B. SDH-deficient RCC with loss of SDHB expression on immunostaining. The stromal cells serve as an internal control and retain their normal staining pattern (immunoperoxidase, 100x).
Hyperparathyroidism-jaw tumor syndrome (HPT-JT) is a rare autosomal dominantly inherited disorder caused by a mutation of the cell division cycle protein 73 homolog (CDC73) gene, also known as the hyperparathyroidism (HRPT2) gene on chromosome 1q24-32 [62, 63]. CDC73 encodes the parafibromin protein, which interacts with β-catenin to form part of the RNA polymerase-associated factor-1 complex. This complex functions as an inhibitor of the c-Myc proto-oncogene [64, 65]. Patients have a predisposition to developing solitary or multiple parathyroid adenomas, parathyroid carcinoma, and multiple ossifying jaw fibromas. Benign uterine tumors (adenofibromas and leiomyomas) and malignant uterine tumors (adenosarcomas) have been reported in up to 75% of female patients [66]. Cystic kidney disease is the most common renal manifestation of HPT-JT. However, in addition to renal hamartomas, these patients are at an increased risk of developing multiple, bilateral renal tumors (approximately 15%), particularly adult Wilms tumors and mixed epithelial-stromal tumors (MEST). Wilms tumors identified in HPT-JT are often diagnosed in the fifth decade of life and usually do not metastasize. Grossly, they are often smaller in size compared to the childhood form of Wilms tumor and poorly circumscribed. Moreover, compared to the childhood form of Wilms tumor, the adult form presenting in HPT-JT shows fewer mitoses, a lack of necrosis and hemorrhage, large mesenchymal components, and cysts [66, 67]. MESTs are characterized by a biphasic growth pattern, including epithelial and spindle cell stromal elements with the spindle cell component often resembling ovarian stroma. These tumors may show sarcomatoid differentiation and undergo malignant transformation with metastasis. Rare cases of papillary RCC have also been reported [68]. As these patients often present with multiple, bilateral renal tumors, they may require multiple surgeries over their lifetime. Nephron-sparing surgery is preferred over radical nephrectomy to preserve long-term kidney function.
Rare cases of balanced translocations of chromosome 3 leading to familial RCC have been described [69-71] and may be due to a loss of the rearranged chromosome during mitosis. Because multiple genes involved in the pathogenesis of renal malignancies are located on chromosome 3 (particularly those with clear cell histology), this type of alteration may confer an increased risk of renal cancer.
Somatic BAP1 mutations have been identified in RCCs and are associated with higher tumor grade and decreased overall survival [72]. However, recent studies have identified germline mutations of BAP1 that predispose to a familial type of clear cell RCC, as well as uveal and cutaneous melanomas, and mesothelioma [73, 74]. BAP1 is a tumor suppressor gene encoding nuclear deubiquitinase, which removes ubiquitin from transcriptional regulator host cell factor (HCF) 1. HCF1 is important for chromatin remodeling, E2F family transcription factor regulation, and downstream cell cycle regulation [75-77]. Penetrance is high and approximately 85% of individuals develop at least one tumor, most are melanocytic, and approximately 10-12% of individuals develop RCCs that are multifocal, bilateral, and occur at a younger age than sporadic RCCs [78]. Given that such cases are rare, evaluation of BAP1 mutation is not routinely performed in cases of inherited RCC and no agreed-upon surveillance protocols for BAP1 mutation carriers exist. Morphologically, these tumors frequently demonstrate features similar to conventional ccRCC combined with a distinct pattern of anastomosing elongated tubules lined by clear cells (Figure 5A) with notably voluminous and partially granular eosinophilic cytoplasm, large cytoplasmic globules, and high-grade nuclear features. Loss of nuclear BAP1 (Figure 5B) is seen in most tumors. As reported in a recent study, these tumors often strongly express p504S, a feature not typically seen in clear cell RCCs [78]. The combination of strong p504S immunoreactivity and loss of BAP1 nuclear staining in tumors morphologically suspicious for this entity, particularly in cases with an established diagnosis of clear cell RCC, should prompt an evaluation for BAP1 mutations molecular testing. These tumors often extend beyond the kidney (pT3), show a high rate of renal vein involvement, and are highly associated with distant metastatic disease.
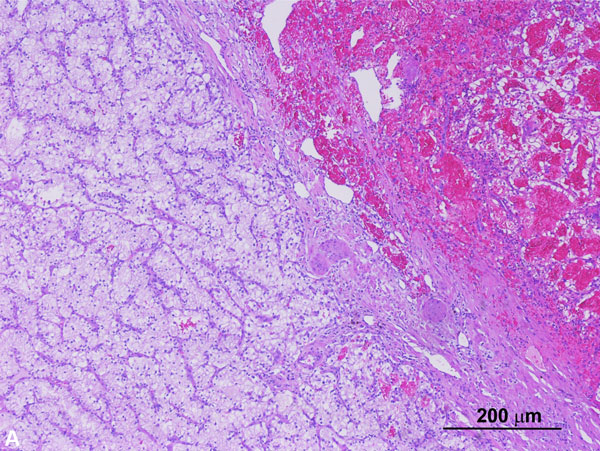
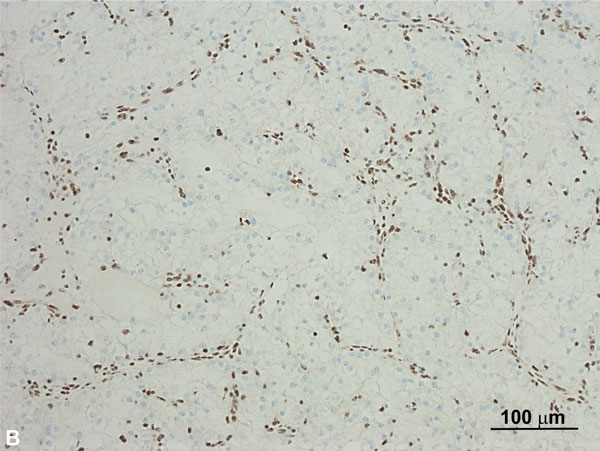
Figure 5A. BAP-1 mutated clear cell RCC. The right half of the tumor in this low-power image shows features similar to conventional clear cell RCC whereas the left half shows a distinct pattern of anastomosing elongated tubules lined by clear cells (H and E, 40x). Figure 5B. BAP1 mutated clear cell RCC with loss of BAP1in tumor cells (Immunoperoxidase, 100x). The stromal and inflammatory cells represent internal controls and show no loss of normal expression.
PTEN hamartoma syndrome encompasses multiple hamartoma syndromes, including Cowden syndrome, Bannayan-Riley-Ruvalcaba syndrome, PTEN-related Proteus syndrome, and PTEN-related Proteus-like syndrome. Specifically, Cowden syndrome is due to autosomal dominantly inherited mutations in PTEN and is associated with an increased risk of benign and malignant tumors of the breast, thyroid, endometrium, and kidney. Skin manifestations of Cowden syndrome include trichilemmomas, papillomatous papules, and acral and plantar keratosis. The most frequently reported renal malignancies in patients with Cowden syndrome include pRCC (72%), chRCC (18%), and ccRCC (9%) with a lifetime risk of 34% and a rising risk beginning at age 40 [79-81]. However, the role of PTEN alterations and behavior in patients with RCC is not well understood. Morphologically, these tumors exhibit papillary, tubular, or solid patterns with fibrovascular cores and foamy macrophages resembling pRCC, solid growth patterns with oncocytic morphology resembling chRCC, or solid and nested patterns with intricate, branching vasculature, and clear cells resembling ccRCC.
a. VHL syndrome
Surgical treatment with total or partial nephrectomy remains the standard of care for localized kidney tumors with a solid tumor component exceeding 3 cm. Partial nephrectomy for relatively smaller and/or circumscribed tumors helps preserve renal parenchyma [82]. Repeat surgical interventions are mandated in recurrent kidney tumors [83]. Anti-VEGF tyrosine kinase inhibitors are used to treat localized germline VHL-associated ccRCC and have demonstrated variable efficacy in these cases [84-86].
b. Hereditary papillary renal cell carcinoma
Renal tumors in hereditary pRCC cases are monitored until they exceed 3 cm [87]. Clinical trials using MET pathway inhibitors to treat hereditary pRCC patients with advanced disease and activating MET mutations are underway. Foretinib, a kinase inhibitor of MET and VEGFR2, has been used to treat patients with pRCC; a small study of ten cases showing partial response suggests it is helpful in cases of germline pathogenic MET variants [88].
c. HLRCC- associated renal cell carcinoma
HLRCC- associated RCCs are aggressive tumors that are often metastatic at the time of diagnosis. These tumors should be treated promptly with complete excision and are exempt from any monitoring size criteria upon recognition. There are no definitive protocols for management of metastatic disease. However, in addition to agents already approved for all RCCs, several clinical trials are investigating optimal treatment regimens. A combination of bevacizumab and erlotinib has been proposed, as combined VEGF and epidermal growth factor receptor blockade would impair blood flow and glucose delivery to the tumor. This regimen can achieve an objective response and is listed for non-ccRCC in the National Comprehensive Cancer Network (NCCN) guidelines [89].
d. Birt-Hogg-Dube syndrome
Oncocytic tumors and chRCCs in BHD syndrome are usually indolent when they are relatively small lesions that can be managed with partial nephrectomy or a nephron-sparing approach if they exceed a size cutoff of 3 cm. Metastases are uncommon but may be seen in tumors with clear cell or papillary features. In a large series of 115 cases of germline FLCN carriers, 14 patients who were not undergoing surveillance developed RCCs, and 5 showed metastatic disease [90]. There is no well-defined systemic treatment approach for these tumors as there are very few cases of metastatic disease.
e. Tuberous sclerosis complex (TSC)-associated kidney tumors
Definitive management in this category depends on tumor size and histological subtyping. Surveillance is performed in cases of lipid-rich or biopsy-proven lipid-poor AMLs until the largest tumor exceeds 4 cm in maximum dimension. Angioembolization may be considered to decrease the risk for spontaneous hemorrhage, but these tumors can be treated by radiologic surveillance in many cases, even when they exceed a maximum size of 4 cm. All malignant tumors are surgically resected. Everolimus is a Food and Drug Administration (FDA)-approved drug for TSC-related AML treatment. A randomized, placebo-controlled EXIST-II trial assessed the efficacy of everolimus in 118 patients with AML and the targeted response rate (50% or more reduction in the total volume of all target AMLs) was 42% for everolimus versus an absent response for placebo (p < .001). However, re-growth of tumor occurred if everolimus was discontinued [91].
f. Succinate dehydrogenase (SDH)-deficient renal cell carcinoma
Cases of RCC arising in patients with SDH germline pathogenic variants are managed similarly to HLRCC, with surgical resection and aggressive treatment strategies, as even small tumors may be metastatic in this cohort of RCCs. Metastatic cases are managed similarly to other RCCs and efforts to identify novel therapeutic targets are ongoing. Inhibitors of glucose uptake, glycolysis, and fatty acid synthesis represent a potential treatment strategy for managing metastatic SDH-deficient and HLRCC-associated RCCs [92]. A current clinical trial is investigating whether vandetanib in combination with metformin can manage these RCCs (NCT02495103).
g. BAP1 tumor predisposition syndrome
BAP1-associated RCC is managed similarly to sporadic RCC, although cases are associated with aggressive behavior or poor outcomes and early intervention with screening is recommended [93]. There are no evidence-based guidelines for cancer screening in individuals with germline BAP1 pathogenic variants, though annual abdominal imaging to screen for RCC has been suggested.
h. Constitutional chromosomal 3 translocations, PTEN hamartoma/Cowden syndrome, and HPT-JT syndrome
As of this writing, NCCN does not include specific management guidelines for renal cancers associated with constitutional chromosomal 3 translocations, PTEN hamartoma/Cowden syndrome, and HPT-JT syndrome. One recently published guideline recommends ultrasound surveillance for RCC in patients with PTEN hamartoma/Cowden syndrome [94]. Other than previously described syndromes, several familial cohorts with multiple cases of RCC without other syndromic features or associated germline mutations have been identified as well. Because of their paucity and lack of strong evidence, it is reasonable to follow the management and surveillance guidelines published for sporadic counterparts until further evidence becomes available. However, such management should be individualized in the light of each tumor’s presentation and behavior. In addition, these cases should be enrolled in pertinent clinical studies whenever possible.
Though only a small subset of RCCs occur as a result of hereditary susceptibility, recognizing these individuals and their family members is important. In the setting of early onset, bilateral, or multicentric renal tumors with appropriate histopathological findings, the possibility of a hereditary etiology should be considered and prompt further investigation through molecular testing. Through early recognition and detection of tumors at a lower stage, the morbidity and mortality of these patients can effectively be reduced either through monitoring of smaller renal lesions in regular intervals, or prompt ablative or surgical approaches as necessary.
Acknowledgements
The authors thank Dr. Heather McConnell and Dr. Sasha Pejerrey for their excellent editorial work.
Ethical policy
This review article was waived from IRB approval.
Author contributions
JYR, Study design; HS and DB, literature research; HS, MKD, DB and JYR, manuscript preparation; MKD, provided photos; MKD, DB and JYR, manuscript review and proof reading; HS, MKD, DB, IYR, all authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Funding
This study was financially not supported by any Institution.
- Moch H, Amin MB, Argani P, Cheville J, Delahunt B, Martignoni G, Medeiros LJ, Srigley JR, Tan PH, Tickoo SK: Renal cell tumors Introduction. In: Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM. WHO Classification of Tumours of the Urinary System and Male Genital Organs. Lyon, France: International Agency for Research on Cancer 2016: p14-17.
- Maher ER, Yates JR, Harries R, Benjamin C, Harris R, Moore AT, Ferguson-Smith MA: Clinical features and natural history of von Hippel–Lindau disease. Q J Med 1990, 77: 1151–1163.
- Gossage L, Eisen T, Maher ER: VHL. The story of a tumour suppressor gene. Nat Rev Cancer 2015, 15: 55–64.
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ: The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399: 271–275.
- Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG, Jr: Inhibition of HIF is necessary for tumor suppression by the von Hippel–Lindau protein. Cancer Cell 2002, 1: 237–246.
- Schödel J, Grampp S, Maher ER, Moch H, Ratcliffe PJ, Russo P, Mole DR: Hypoxia, hypoxia-inducible transcription factors, and renal cancer. Eur Urol 2016, 69: 646–657.
- Ong KR, Woodward ER, Killick P, Lim C, Macdonald F, Maher ER: Genotype–phenotype correlations in von Hippel–Lindau disease. Hum Mutat 2007, 28: 143–149.
- Zbar B, Kishida T, Chen F, Maher ER, Richards FM, Crossey PA, Webster AR, Affara NA, Ferguson-Smith MA et al: Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat 1996, 8: 348-357.
- Hes F, Zewald R, Peeters T, Links T, Verheij J, Matthijs G, Leguis E, Mortier G, van der Torren K et al: Genotype-phenotype correlations in families with deletions in the von Hippel-Lindau (VHL) gene. Hum Genet 2000, 106: 425-431.
- Chen F, Kishida T, Yao M, Hustad T, Glavac D, Dean M, Gnarra JR, Orcutt ML, Duh FM, Glenn G, et al: Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat 1995, 5: 66-75.
- Crossey PA, Foster K, Richards FM, Phipps ME, Latif F, Tory K, Jones MH, Bentley E, Kumar R, Lerman MI et al: Molecular genetic investigations of the mechanism of tumourigenesis in von Hippel-Lindau disease: analysis of allele loss in VHL tumours. Hum Genet 1994, 93: pp. 53-58.
- Banks RE, Tirukonda P, Taylor C, Hornigold N, Astuti D, Cohen D, Maher ER, Stanley AJ, Harnden P, Joyce A et al: Genetic and epigenetic analysis of von Hippel-Lindau (VHL) gene alterations and relationship with clinical variables in sporadic renal cancer. Cancer Res 2006, 66: 2000-2011.
- Dondeti VR, Wubbenhorst B, Lal P, Gordan JD, D'Andrea K, Attiyeh EF, Simon MC, Nathanson KL: Integrative genomic analyses of sporadic clear cell renal cell carcinoma define disease subtypes and potential new therapeutic targets. Cancer Res 2012, 72: 112-121.
- Gordan JD, Lal P, Dondeti VR, Letrero R, Parekh KN, Oquendo CE, Greenberg RA, Flaherty KT, Rathmell WK, Keith B et al: HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell 2008, 14: 435-446.
- Walther MM, Choyke PL, Glenn G, Lyne JC, Rayford W, Venzon D, Linehan WM: Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J Urol 1999, 161: 1475-1479.
- Solomon D, Schwartz A: Renal pathology in von Hippel-Lindau disease. Hum Pathol 1988, 19: 1072-1079.
- Chen YB, Tickoo SK: Spectrum of preneoplastic and neoplastic cystic lesions of the kidney. Arch Pathol Lab Med 2012, 136: 400-409.
- Williamson SR, Zhang S, Eble JN, Grignon DJ, Martignoni G, Brunelli M, Wang M, Gobbo S, Baldridge LA, Cheng L: Clear cell papillary renal cell carcinoma-like tumors in patients with von Hippel-Lindau disease are unrelated to sporadic clear cell papillary renal cell carcinoma. Am J Surg Pathol 2013, 37: 1131-1139.
- Rao P, Monzon F, Jonasch E, Matin SF, Tamboli P: Clear cell papillary renal cell carcinoma in patients with von Hippel-Lindau syndrome--clinicopathological features and comparative genomic analysis of 3 cases. Hum Pathol 2014, 45: 1966-1972.
- Schimdt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P, Scherer SW, Zhuang Z, Lubensky I, Dean M et al: Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet 1997, 16: 68–73.
- Lubensky IA, Schmidt L, Zhuang Z, Weirich G, Pack S, Zambrano N, Walther MM, Choyke P, Linehan WM, Zbar B: Hereditary and sporadic papillary renal carcinomas with c-met mutations share a distinct morphological phenotype. Am J Pathol 1999, 155: 517-526.
- Tovar EA, Graveel CR: MET in human cancer: germline and somatic mutations. Ann Transl Med 2017, 5(10): 205.
- Peng YC, Chen YB: Recognizing hereditary renal cancers through the microscope: A Pathology Update. Surg Pathol Clin 2018, 11: 725-737.
- Maher ER: Hereditary renal cell carcinoma syndromes: diagnosis, surveillance and management. World J Urol. 2018, 36: 1891-1898.
- Lehtonen HJ: Hereditary leiomyomatosis and renal cell cancer: update on clinical and molecular characteristics. Fam Cancer 2011, 10: 397–411.
- Hansen AW, Chayed Z, Pallesen K, Codruta Vasilescu I, Bygum A: Hereditary Leiomyomatosis and Renal Cell Cancer. Acta Derm Venereol. 2020; 100:adv00012.
- Brown S, Brennan P, Rajan N: Inherited skin tumour syndromes. Clin Med (Lond) 2017, 17: 562–567.
- Tomlinson IPM, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S et al: Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 2002, 30: 406-410.
- Chayed Z, Kristensen LK, Ousager LB, Rønlund K, Bygum A: Hereditary leiomyomatosis and renal cell carcinoma: a case series and literature review. Orphanet J Rare Dis 2021, 16: 34.
- Chen YB, Brannon AR, Toubaji A, Dudas ME, Won HH, Al-Ahmadie HA, Fine SW, Gopalan A, Frizzell N, Voss MH, Russo P et al: Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer: recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am J Surg Pathol. 2014, 38: 627–637.
- Badeloe S, Frank J: Clinical and molecular genetic aspects of hereditary multiple cutaneous leiomyomatosis. Eur J Dermatol 2009, 19: 545-551.
- Toro JR, Nickerson ML, Wei MH, Warren MB, Glenn GM, Turner ML, Stewart L, Duray P, Tourre O, Sharma N et al: Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet 2003, 73: 95-106.
- Wei MH, Toure O, Glenn GM, Pithukpakorn M, Neckers L, Stolle C, Choyke P, Grubb R, Middelton L, Turner ML, Walther MM et al: Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet 2006, 43: 18-27.
- Launonen V, Vierimaa O, Kiuru M, Isola J, Roth S, Pukkala E, Sistonen P, Herva R, Aaltonen LA: Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci USA 2001, 98: 3387-3392.
- Koski TA, Lehtonen HJ, Jee KJ, Ninomiya S, Joosse SA, Vahteristo P, Kiuru M, Karhu A, Sammalkorpi H, Vanharanta S et al: Array comparative genomic hybridization identifies a distinct DNA copy number profile in renal cell cancer associated with hereditary leiomyomatosis and renal cell cancer. Genes Chromosomes Cancer 2009, 48: 544-551.
- Merino MJ, Torres-Cabala C, Pinto P, Linehan WM: The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol 2007, 31: 1578-1585
- Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn G, Turner ML, Duray P, Merino M, Choyke P, Pavlovich CP et al: Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell 2002, 2: 157-164.
- Benhammou JN, Vocke CD, Santani A, Schmidt LS, Baba M, Seyama K, Wu X, Korolevich S, Nathanson KL, Stolle CA et al: Identification of intragenic deletions and duplication in the FLCN gene in Birt-Hogg-Dube syndrome. Genes Chromosomes Cancer 2011, 50: 466-477.
- Luijten MN, Basten SG, Claessens T, Vernooij M, Scott CL, Janssen R, Easton JA, Kamps MA, Vreeburg M, Broers JL et al: Birt-Hogg-Dube syndrome is a novel ciliopathy. Hum Mol Genet 2013, 22: 4383-4397.
- Medvetz DA, Khabibullin D, Hariharan V, Ongusaha PP, Goncharova EA, Schlechter T, Darling TN, Hofmann I, Krymskaya VP, Liao JK et al: Folliculin, the product of the Birt-Hogg-Dube tumor suppressor gene, interacts with the adherens junction protein p0071 to regulate cell-cell adhesion. PLoS One 2012, 7: e47842.
- Gaur K, Li J, Wang D, Yan SJ, Tsurumi A, Land H, Wu G, Li WX: The Birt-Hogg-Dube tumor suppressor Folliculin negatively regulates ribosomal RNA synthesis. Hum Mol Genet 2013, 22:284-299.
- Schmidt LS, Linehan WM: FLCN: The causative gene for Birt-Hogg-Dubé syndrome. Gene 2018, 640: 28-42.
- Muller ME, Daccord C, Taffé P, Lazor R: Prevalence of Birt-Hogg-Dubé syndrome determined through epidemiological data on spontaneous pneumothorax and Bayes theorem. Front Med (Lausanne). 2021, 8: 631168.
- Khoo SK, Bradley M, Wong FK, Hedblad MA, Nordenskjold M, Teh BT: Birt-Hogg-Dube syndrome: mapping of a novel hereditary neoplasia gene to chromosome 17p12-q11.2. Oncogene 2001, 20: 5239-5242.
- Schmidt LS, Warren MB, Nickerson ML, Weirich G, Matrosova V, Toro JR, Turner ML, Duray P, Merino M, Hewitt S et al: Birt-Hogg-Dube syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet 2001, 69: 876-882.
- Pfaffenroth EC, Linehan WM: Genetic basis for kidney cancer: opportunity for disease-specific approaches to therapy. Expert Opin Biol Ther 2008, 8: 779-790.
- Toro JR, Wei MH, Glenn GM, Weinreich M, Toure O, Vocke C, Turner M, Choyke P, Merino MJ, Pinto PA et al: BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet 2008, 45: 321-331.
- Kluijt I, de Jong D, Teertstra HJ, Axwijk PH, Gille JJ, Bell K, van Rens A, van der Velden AW, Middelton L, Horenblas S et al: Early onset of renal cancer in a family with Birt-Hogg-Dubé syndrome. Clin Genet 2009, 75: 537-543.
- Pavlovich CP, Walther MM, Eyler RA. Hewitt SM, Zbar B, Linehan WM, Merino MJ: Renal tumors in the Birt-Hogg-Dubé syndrome. Am J Surg Pathol 2002, 26: 1542-1552.
- Pavlovich CP, Schmidt LS: Searching for the hereditary causes of renal-cell carcinoma. Nature Rev 2004, 4: 381-393.
- Pavlovich CP, Grubb RL, Hurley K, Glenn GM, Toro J, Schmidt LS, Torres-Cabala C, Merino MJ, Zbar B, Choyke P et al: Evaluation and management of renal tumors in the Birt-Hogg-Dube syndrome. J Urol 2005, 173: 1482-1486.
- Menko FH, van Steensel MA, Giraud S, Friis-Hansen L, Richard S, Ungari S, Nordenskjöld M, Hansen TV, Solly J, Maher ER: European BHD Consortium: Birt-Hogg-Dube syndrome: diagnosis and management. Lancet Oncol 2009, 10: 1199-1206.
- Peng YC, Chen YB: Recognizing Hereditary Renal Cancers Through the Microscope: A Pathology Update. Surg Pathol Clin. 2018, 11: 725-737.
- Crino PB, Nathanson KL, Henske EP: The tuberous sclerosis complex. N Engl J Med 2006, 355: 1345-1356.
- Kozlowski P, Roberts P, Dabora S, Franz D, Bissler J, Northrup H, Au KS, Lazarus R, Domanska-Pakiela D, Kotulska K et al: Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet 2007, 121: 389-400.
- Dixon BP, Hulbert JC, Bissler JJ: Tuberous sclerosis complex renal disease. Nephron Exp Nephrol 2011, 118: e15-e20.
- Yang P, Cornejo KM, Sadow PM, Cheng L, Wang M, Xiao Y, Jiang Z, Oliva E, Jozwiak S, Nussbaum RL et al: Renal cell carcinoma in tuberous sclerosis complex. Am J Surg Pathol. 2014, 38: 895-909.
- Gill AJ, Hes O, Papathomas T, Šedivcová M, Tan PH, Agaimy A, Andresen PA, Kedziora A, Clarkson A, Toon CW et al: Succinate dehydrogenase (SDH)-deficient renal carcinoma: a morphologically distinct entity: a clinicopathologic series of 36 tumors from 27 patients. Am J Surg Pathol 2014, 38: 1588-1602.
- Gill AJ: Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathology 2012, 44: 285-292.
- Ricketts C, Woodward ER, Killick P, Morris MR, Astuti D, Latif F, Maher ER: Germline SDHB Mutations and Familial Renal Cell Carcinoma. J Natl Cancer Inst 2008, 100: 1260–1262.
- Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peçzkowska M, Morrison CD, Lehtonen R, Januszewicz A, Järvinen H, Juhola M: Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet 2004, 74: 153–159.
- Pavlovich CP, Schmidt LS: Searching for the hereditary causes of renal-cell carcinoma. Nature Rev 2004, 4: 381-393.
- Newey PJ, Bowl MR, Thakker RV: Parafibromin—functional insights. J Intern Med 2009, 266: 84-98.
- Lin L, Zhang JH, Panicker LM, Simonds WF: The parafibromin tumor suppressor protein inhibits cell proliferation by repression of the c-Myc proto-oncogene. Proc Natl Acad Sci USA 2008, 105: 17420-17425.
- Verine J, Pluvinage A, Bousquet G, Lehmann-Che J, de Bazelaire C, Soufir N, Mongiat-Artus P. Hereditary renal cancer syndromes: an update of a systematic review. Eur Urol 2010, 58: 701-710.
- Szabó J, Heath B, Hill VM, Jackson CE, Zarbo RJ, Mallette LE, Chew SL, Besser GM, Thakker RV, Huff V et al: Hereditary hyperparathyroidism-jaw tumor syndrome: the endocrine tumor gene HRPT2 maps to chromosome 1q21-q31. Am J Hum Genet 1995, 56: 944-950.
- Teh BT, Farnebo F, Kristoffersson U, Sundelin B, Cardinal J, Axelson R, Yap A, Epstein M, Heath H 3rd, Cameron D et al: Autosomal dominant primary hyperparathyroidism and jaw tumor syndrome associated with renal hamartomas and cystic kidney disease: linkage to 1q21-q32 and loss of the wild type allele in renal hamartomas, J Clin Endocrinol Metab 1996, 81: 4204-4211.
- Haven CJ, Wong FK, van Dam EW, van der Juijt R, van Asperen C, Jansen J, Rosenberg C, de Wit M, Roijers J, Hoppener J et al: A Genotypic and Histopathological Study of a Large Dutch Kindred with Hyperparathyroidism-Jaw Tumor Syndrome. J Clin Endocrinol Metab 2000, 85: 1449–1454.
- Bodmer D, Eleveld M, Ligtenberg M, Weterman M, van der Meijden A, Koolen M, Hulsbergen-van der Kaa C, Smits A, Smeets D, Geurts van Kessel A: Cytogenetic and molecular analysis of early stage renal cell carcinomas in a family with a translocation (2;3) (q35;q21). Cancer Genet Cytogenet 2002, 134: 6-12.
- Bonne AC, Bodmer D, Schoenmakers EF, van Ravenswaaij CM, Hoogerbrugge N, van Kessel AG: Chromosome 3 translocations and familial renal cell cancer. Curr Mol Med 2004, 4: 849-854.
- Kanayama H, Lui WO, Takahashi M, Naroda T, Kedra D, Wong FK, Kuroki Y, Nakahori Y, Larsson C, Kagawa S et al: Association of a novel constitutional translocation t(1q;3q) with familial renal cell carcinoma. J Med Genet 2001, 38: 165-170.
- Kapur P, Pena-Llopis S, Christie A, Zhrebker L, Pavía-Jiménez A, Rathmell WK, Xie XJ, Brugarolas J: Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: a retrospective analysis with independent validation. Lancet Oncol 2013, 14: 159-167.
- Farley MN, Schmidt LS, Mester JL, Pena-Llopis S, Pavia-Jimenez A, Christie A, Vocke CD, Ricketts CJ, Peterson J, Middelton L et al: A novel germline mutation in BAP1 predisposes to familial clear-cell renal cell carcinoma. Mol Cancer Res 2013, 11: 1061-1071.
- Popova T, Hebert L, Jacquemin V, Gad S, Caux-Moncoutier V, Dubois-d'Enghien C, Richaudeau B, Renaudin X, Sellers J, Nicolas A et al: Germline BAP1 mutations predispose to renal cell carcinomas. Am J Hum Genet 2013, 92: 974-980.
- Fukuda T, Tsuruga T, Kuroda T, Nishikawa H, Ohta T: Functional link between BRCA1 and BAP1 through histone H2A, heterochromatin and DNA damage response. Curr Cancer Drug Targets 2016, 16: 101-109.
- Wang SS, Gu YF, Wolff N, Stefanius K, Christie A, Dey A, Hammer RE, Xie XJ, Rakheja D, Pedrosa I et al: Bap1 is essential for kidney function and cooperates with Vhl in renal tumorigenesis. Proc Natl Acad Sci USA 2014, 111: 16538-16543.
- Jin S, Wu J, Zhu Y, Gu W, Wan F, Xiao W, Dai B, Zhang H, Shi G, Shen Y et al: Comprehensive analysis of BAP1 somatic mutation in clear cell renal cell carcinoma to explore potential mechanisms in silico. J Cancer 2018, 9: 4108-4116.
- Gallan AJ, Parilla M, Segal J, Ritterhouse L, Antic T: BAP1-Mutated Clear Cell Renal Cell Carcinoma: A Clinicopathologic Characterization of 14 Molecularly Confirmed Tumors, Am J Clini Pathol 2021, 155: 718–728.
- Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 2012, 18: 400-407.
- Haibach, H., Burns, TW, Carlson HE, Burman KD, Deftos LJ: Multiple hamartoma syndrome (Cowden’s disease) associated with renal cell carcinoma and primary neuroendocrine carcinoma of the skin (Merkel cell carcinoma). Am J Clin Pathol 1992, 97: 705–712.
- Kim RH, Wang X, Evans AJ, Campbell SC, Nguyen JK, Farncombe KM, Eng C: Early-onset renal cell carcinoma in PTEN harmatoma tumour syndrome. NPJ Genom. Med 2020, 5: 40
- Duffey BG, Choyke PL, Glenn G, Grubb RL, Venzon D, Linehan WM, Walther MM: The relationship between renal tumor size and metastases in patients with von Hippel-Lindau disease. J Urol 2004, 172: 63–65.
- Singer EA, Vourganti S, Lin KY, Gupta GN, Pinto PA, Rastinehad AR, Linehan WM, Bratslavsky G: Outcomes of patients with surgically treated bilateral renal masses and a minimum of 10 years of follow up. J Urol 2012, 188: 2084–2088.
- Jonasch E, McCutcheon IE, Waguespack SG, Wen S, Davis DW, Smith LA, Tannir NM, Gombos DS, Fuller GN, Matin SF: Pilot trial of sunitinib therapy in patients with von Hippel-Lindau disease. Ann Oncol 2011, 22: 2661–2666.
- Kim HC, Lee JS, Kim SH, So HS, Woo CY, Lee JL. Sunitinib treatment for metastatic renal cell carcinoma in patients with von Hippel- Lindau disease. Cancer Res Treat 2013, 45: 349–353.
- Jonasch E, McCutcheon IE, Gombos DS, Ahrar K, Perrier ND, Liu D, Robichaux CC, Villarreal MF, Weldon JA, Woodson AH et al: Pazopanib in patients with von Hippel-Lindau disease: a single-arm, single-centre, phase 2 trial. Lancet Oncol 2018, 19: 1351–1359.
- Sulkowski PL, Sundaram RK, Oeck S, Corso CD, Liu Y, Noorbakhsh S, Niger M, Boeke M, Ueno D, Kalathil AN et al: Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat Genet 2018, 50: 1086–1092.
- Choueiri TK, Vaishampayan U, Rosenberg JE, Logan TF, Harzstark AL, Bukowski RM, Rini BI, Srinivas S, Stein MN, Adams LM et al: Phase II: and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol 2013, 31: 181–186.
- Srinivasan R, Gurram S, Harthy MA, Singer EA, Sidana A, Shuch BM, Ball MW, Friend JC, Mac L, Purcell E et al: Results from a phase II study of bevacizumab and erlotinib in subjects with advanced hereditary leiomyomatosis and renal cell cancer (HLRCC) or sporadic papillary renal cell cancer. Journal of Clinical Oncology 2020, 38(15_suppl): 5004-5004.
- Schmidt LS, Linehan WM: Molecular genetics and clinical features of Birt-Hogg-Dube syndrome. Nat Rev Urol 2015, 12: 558–569.
- Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA, Frost M, Belousova E, Sauter M, Nonomura N, Brakemeier S, de Vries PJ et al: Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2013, 381: 817–824.
- Lee C-H, Gundem G, Lee W, Chen Y-B, Cross JR, Dong Y, Redzematovic A, Mano R, Wei EY, Cheng EH et al: Persistent Severe Hyperlactatemia and Metabolic Derangement in Lethal SDHB-Mutated Metastatic Kidney Cancer: Clinical Challenges and Examples of Extreme Warburg Effect. JCO Precision Oncology 2017, https://doi.org/10.1200/po.16.00007. Epub ahead of print.(1):1-14.
- Shuch B, Zhang J. Genetic predisposition to renal cell carcinoma implications for counseling, testing, screening, and management. J Clin Oncol 2018, 36: 3560–3569.
- Tischkowitz M, Colas C, Pouwels S, Hoogerbrugge N: Cancer Surveillance Guideline for individuals with PTEN hamartoma tumour syndrome. Eur J Hum Genet. 2020, 28: 1387-1393.
- Rednam SP, Erez A, Druker H, Janeway KA, Kamihara J, Kohlmann WK, Nathanson KL, States LJ, Tomlinson GE, Villani A et al: Von Hippel-Lindau and Hereditary Pheochromocytoma/Paraganglioma Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin Cancer Res 2017, 23(12): e68-e75.
- Schultz KAP, Rednam SP, Kamihara J, Doros L, Achatz MI, Wasserman JD, Diller LR, Brugieres L, Druker H, Schneider KA et al: PTEN, DICER1, FH, and Their Associated Tumor Susceptibility Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin Cancer Res 2017, 23(12): e76-e82.
- Stamatakis L, Metwalli AR, Middelton LA, Marston Linehan W: Diagnosis and management of BHD-associated kidney cancer. Fam Cancer 2013, 12(3): 397-402.
- Krueger DA, Northrup H, International Tuberous Sclerosis Complex Consensus G: Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 2013, 49(4): 255-265.
- Hyde SM, Rich TA, Waguespack SG, Perrier ND, Hu MI: CDC73-Related Disorders. In: GeneReviews(®). Epub ahead of print., edn. Edited by Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A. Seattle (WA): University of Washington, Seattle.
- Star P, Goodwin A, Kapoor R, Conway RM, Long GV, Scolyer RA, Guitera P: Germline BAP1-positive patients: the dilemmas of cancer surveillance and a proposed interdisciplinary consensus monitoring strategy. Eur J Cancer 2018, 92: 48-53.
- Daly MB, Pilarski R, Berry M, Buys SS, Farmer M, Friedman S, Garber JE, Kauff ND, Khan S, Klein C et al: NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J Natl Compr Canc Netw 2017, 15(1): 9-20.
Annals of urologic oncology
p-ISSN: 2617-7765, e-ISSN: 2617-7773
 Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.