
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
Research Progress of AR Gene Family in Prostate Cancer Therapy
Maham Khan1
1Pharmacokinetics Department, Aston University, Birmingham B4 7ET, England, United Kingdom.
Correspondence: Maham Khan (Pharmacokinetics Department, Aston University, Birmingham B4 7ET, England, United Kingdom; Email: mahamkhan3692@gmail.com).
Annals of Urologic Oncology 2024, 7(4): 177-184. https://doi.org/10.32948/auo.2024.10.13
Received: 06 Oct 2024 | Accepted: 13 Oct 2024 | Published online: 18 Oct 2024
Key words prostate cancer, AR family, AKR1C3, AR-V7, tumor microenvironment
AKR1C3 belongs to the aldoketone reductase superfamily and like other AKR enzymes, it is a soluble monomer enzyme that depends on NAD(P)(H) to catalyze the reduction of aldehydes and ketones. Studies have shown that AKR1C3 is significantly expressed in metastatic and recurrent prostate xenografts and bone metastases in CRPC patients [8, 9], highlighting its role in androgen synthesis. AKR1C3 is a key enzyme in the testosterone and dihydrotestosterone synthesis pathway [10]. AKR1C3 is a key enzyme that promotes the production of androgens within tumor cells, an adaptive mechanism to maintain their survival after serum testosterone levels. In addition, overexpression of AKR1C3 in prostate cancer cells is associated with enhanced intra-tumor androgen synthesis [11], even in the context of ADT. This intracellular androgen production allows tumor cells to evade systemic androgen deprivation and continue to grow and progress, leading to the development of CRPC phenotype [12]. As a result, targeting AKR1C3 has emerged as a potential therapeutic strategy to inhibit intra-tumor androgen synthesis and combat resistance to ADT in prostate cancer.
Moreover, prostate cancer patients with high levels of AKR1C3 [17] expression are often resistant to radiotherapy and exhibit resistance to anti-androgen therapies such as enzalutamide (Enza). Recent studies, however, have indicated that AKR1C3 functions as a selective coactivator of the androgen receptor (AR) and activates the ERK1/2 pathway, thereby enhancing non-enzymatic mechanisms of prostate metastasis. However, studies on how AKR1C3 promotes malignant progression of prostate cancer through non-enzymatic pathways are still limited [18, 19]. In addition, the ability of prostate cancer cells to produce androgens within the tumor is a key mechanism by which these cells adapt to reduce systemic androgen levels after ADT. This adaptive response not only supports tumor survival, but also drives disease progression. Specifically, the upregulation of enzymes involved in the androgen synthesis pathway, such as AKR1C3, amplifies this process, enabling tumors to maintain a certain level of androgen receptor activation, thereby promoting their growth and metastasis [20]. Therefore, targeting these androgen synthesis pathways has become a promising area of research for developing new therapies that are effective against CRPC.
Previous studies have shown that AR variants (AR-VS) arise from structural rearrangements or selective RNA splicing of AR genes [27]. Selective RNA splicing is the mechanism through which exons are differentially spliced from precursor mRNA (pre-mRNA), leading to the creation of various combinations of exons that ultimately form translatable mRNA. AR-V7 mRNA is selectively spliced at the 3' splice site (3'ss) of exon 3B instead of at the 3' splice site of exon 4. AR-V7 can be regulated by various RNA splicing regulatory factors, such as U2AF65, ASF/SF2 [28], and JMJD1A [29], or through the regulation of cis-acting elements like Hoxb13 [30] and FOXA1 [31]. In addition to transcriptional regulation, the control of AR-V7 protein levels has also been studied. Liu et al. further identified that the HSP70/STUB1 complex mediates AR-V7 protein stabilization, which enhances resistance to next-generation anti-androgen drugs. STUB1 induces AR-V7 protein degradation by blocking the formation of the HSP70/AR-V7 complex [32]. Li et al. reported that PP-1 and Akt signaling regulate AR-V7 serine phosphorylation through the E3 ligase MDM2, which recognizes the phosphorylated sites on AR-V7 and induces its ubiquitination and degradation [33]. Currently, no specific chemotherapeutic agents directly target AR-V7, and the and the available treatments often prove ineffective or show inherent resistance in AR-V7-positive patients. As a result, targeting the epigenetic processes to regulate the production or stability of AR-V7 offers a promising therapeutic strategy for treating patients with AR-V7-positive prostate cancer.
In CRPC 22Rv1 cells, genomic editing of AR gene rearrangements revealed that the stability and high-level expression of truncated AR subtypes are associated with an intragenic rearrangement of a roughly 35 kb AR genomic fragment, which includes AR exon 3 and several 3′-terminal cryptic exons (CE) expressed in AR-Vs, [37]. Subsequently, the same research group discovered an 8579 bp deletion of AR exons 5, 6, and 7 in the LuCaP 86.2 xenograft model, which provided a reasonable explanation for the synthesis of truncated AR-V567 in this model [38]. A study by De Laere, demonstrated that the majority of AR-GSR-positive patients expressed AR-Vs, with only a few exceptions. The association between AR-GSR and AR-Vs has also been confirmed in clinical tumors. AR-GSR in individual tumors showed varying degrees of clonal enrichment, revealing that tumors with high clonal enrichment of AR-GSR were found to express unique AR-GSR-dependent AR-Vs [39]. AR gene rearrangements are an important mechanism for generating AR-Vs in cells that express AR-Vs but not full-length AR (AR-FL).
Recent studies have shown that abnormal RNA intra gene splicing is another key mechanism for AR-Vs expression. This explains why full-length androgen receptor (AR-FL) and AR-Vs can be co-expressed in the same cell in patients with (CRPC) [36]. RNA splicing facilitates mRNA translation by cutting out introns and exons, resulting in multiple transcriptional variants from a single precursor messenger RNA (pre-mRNA). This process allows eukaryotic cells with around 20,000 genes to expand into a diverse proteome containing about 95,000 proteins [40]. mRNA splicing is carried out by the splicosome, a large complex of small nuclear ribonucleoproteins (snRNPs, such as U1, U2, U4, U5, and U6) and their associated helper proteins. The splicosome sequentially binds to the transcribed RNA, recognizing specific sequence elements in the intron (such as the 5' splicing site and the 3' splicing site) to cut out the intron in the pre-mRNA and link the adjacent exons together [41]. The expression of specific splicing variants is regulated by the rate of gene transcription and the splicing factors that bind to pre-mRNA during splicing [42]. Both the initiation and elongation rate of transcription can affect splicing. The transcription initiation and elongation rates can influence splicing, while splicing factors regulate the spliceosome’s recognition of splicing sites by binding to regulatory proteins on the pre-mRNA [43]. Enhanced expression of splicing factors promotes their recruitment to pre-mRNA, thereby facilitating this dynamic and reversible mRNA splicing process. These factors bind to specific sequence elements on pre-mRNA, influencing exon inclusion or skipping and thereby regulating the efficiency of the spliceosome's recognition of splicesome [29]. Both AR-V7 and AR-FL are generated through alternative splicing of AR pre-mRNA. AR-V7 is generated by the spliceosome through aberrant splicing at a cryptic exon 3b (3'SS) located in the third intron of AR, rather than at the 3'SS of exon 4 [44]. Numerous splicing factors are involved in the splicing mechanism of AR-V7. Multiple splicing factors are implicated in this splicing mechanism, including PSF/SFPQ, which are rich in proline and glutamine. The expression of PSF/SFPQ has been associated with poor prognosis in prostate cancer patients, as they promote the expression of several spliceosomal genes, leading to increased production of AR-V7.
Currently, chemists are striving to develop selective AKR1C3 inhibitors. In the search of small molecule compounds, such as cinnamic acid derivatives, phenolic acid derivatives, isoquinoline alkaloids, and organic ruthenium complexes, several compounds with IC50 values in the nanomolar range have been identified for AKR1C3 inhibition [48, 49]. However, due to the structural similarities among the aldo-keto reductase family, especially among family members AKR1C1, AKR1C2, and AKR1C4, there remains a need for compounds that are highly selective for AKR1C3 and exhibit favorable pharmacokinetic properties. Novel AKR1C3 inhibitors demonstrating biological efficacy have been developed and are currently undergoing clinical trial [50, 51] (Figure 1).
Among the studied compounds, ASP9521 (3mg/kg) was shown to significantly suppress intratumoral testosterone levels in human CRPC (castration-resistant prostate cancer) xenograft mice with a single oral dose, and this effect persisted for 24 hours [52]. This compound entered phase I/II clinical trials for metastatic CRPC, where it demonstrated acceptable safety and tolerability, though it did not exhibit significant clinical activity, potentially due to suboptimal patient selection [53]. Another inhibitor, BAY1128688, a steroid-based analogue, entered phase II clinical trials for endometriosis but was discontinued due to its association with liver toxicity [54, 55]. Currently, no AKR1C3 inhibitors are specifically approved for prostate cancer treatment. In 2023, HSK38008, a novel oral AR-V7 degrader, was developed, effectively targeting AR-V7 protein degradation through the proteasomal pathway. However, despite these advances, no clinically proven drugs directly inhibit AR-V7 production. Developing such therapies is crucial for overcoming the complexities of androgen receptor signaling in CRPC, offering potential new avenues for managing this challenging disease.
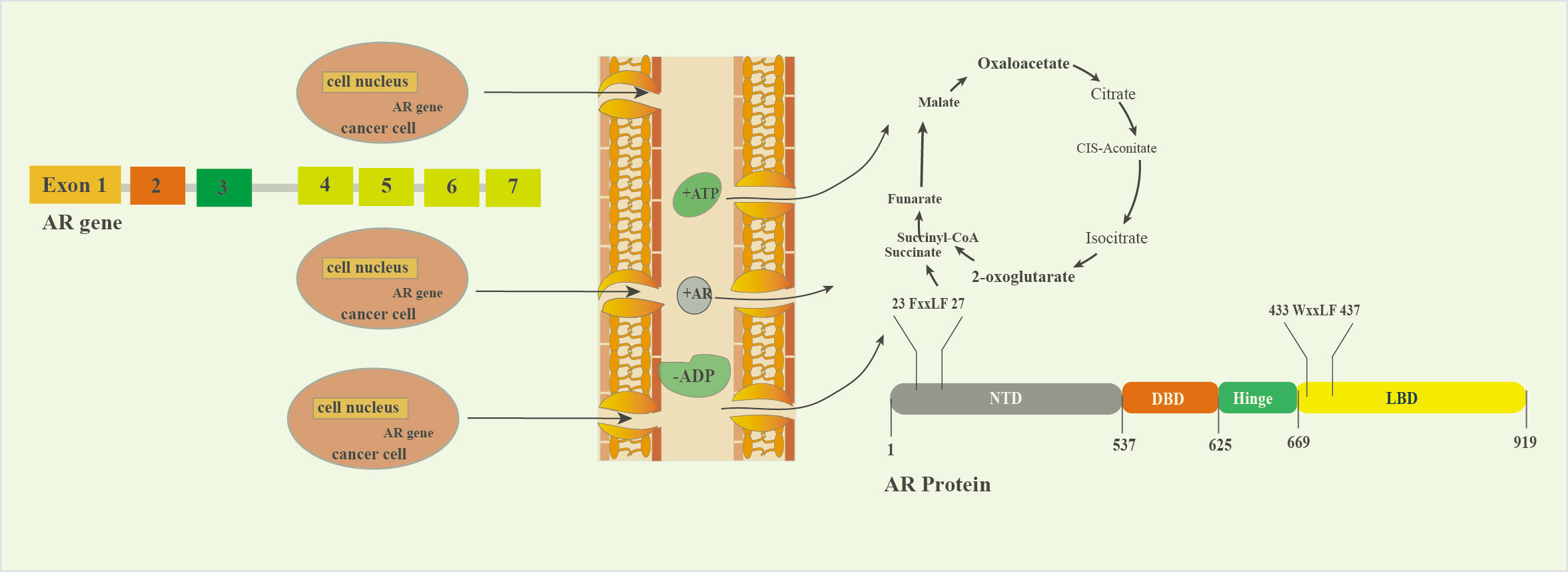 Figure 1. Schematic diagram of AR splicing variants. This diagram illustrates the structural variations of androgen receptor (AR) splice variants. These variants arise from alternative splicing of the AR gene, leading to different protein isoforms that can have distinct biological functions and implications in various conditions, particularly in prostate cancer.
Figure 1. Schematic diagram of AR splicing variants. This diagram illustrates the structural variations of androgen receptor (AR) splice variants. These variants arise from alternative splicing of the AR gene, leading to different protein isoforms that can have distinct biological functions and implications in various conditions, particularly in prostate cancer.
To date over 20 AR variants (AR-Vs) have been identified in human prostate cancer cell models and clinical specimens. These variates arise due to the splicing of "cryptic" exons into upstream exons encoding the AR DNA-binding domain [58]. With the exception of AR45, which is truncated in the N-terminal domain (lacking the entire region encoded by exon 1 of the AR gene) [59], all other variants contain intact N-terminal transactivation and DNA-binding domains but lack the LBD (ligand-binding domain) (Figure 2). Although these truncated proteins cannot bind ligands, they possess constitutive activity as transcription factors and can promote the expression of target genes [60]. As a result, these truncated receptor forms drive androgen-independent transcription of AR target genes, supporting the growth of hormone-independent prostate cancer cells. Enzalutamide which exerts its antitumor activity by interacting with the ligand-binding domain of the androgen receptor, may become less effective in the presence of AR variants, potentially contributing to enzalutamide resistance [61]. Increasing evidence suggests that prostate tumors adapt to androgen deprivation therapies, such as abiraterone and enzalutamide by activating AR target genes through AR-Vs that lack a functional ligand-binding domain.
AR-V7 has been detected in circulating tumor cells (CTCs) isolated from blood samples, whole blood mRNA, ctRNA, tumor-derived cell vesicles, plasma or serum, and even in so-called exosomes found in urine [62-64]. Patients with detectable AR-V7 in liquid biopsies often exhibit more aggressive disease and shorter survival times [65]. Alessandro Sciarra [39] and colleagues analyzed 56 cases of PCa based on clinical risk, detecting AR-V7 expression in 24/32 cases in the high-risk group, 4/13 in the intermediate-risk group, and only 2/11 in the low-risk group. A significant correlation was observed between AR-V7 positivity and risk classification (P<0.001) as well as postoperative progression (P<0.001). AR-V7 has also been identified to predict poor prognosis in mCRPC patients treated with drugs such as enzalutamide or abiraterone [66, 67]. Increasing evidence suggests that alternative splicing pathways play a crucial role in regulating AR-V7 production. Understanding the regulatory mechanisms opens new avenues for prostate cancer treatment strategies focused on targeting AR-V7.
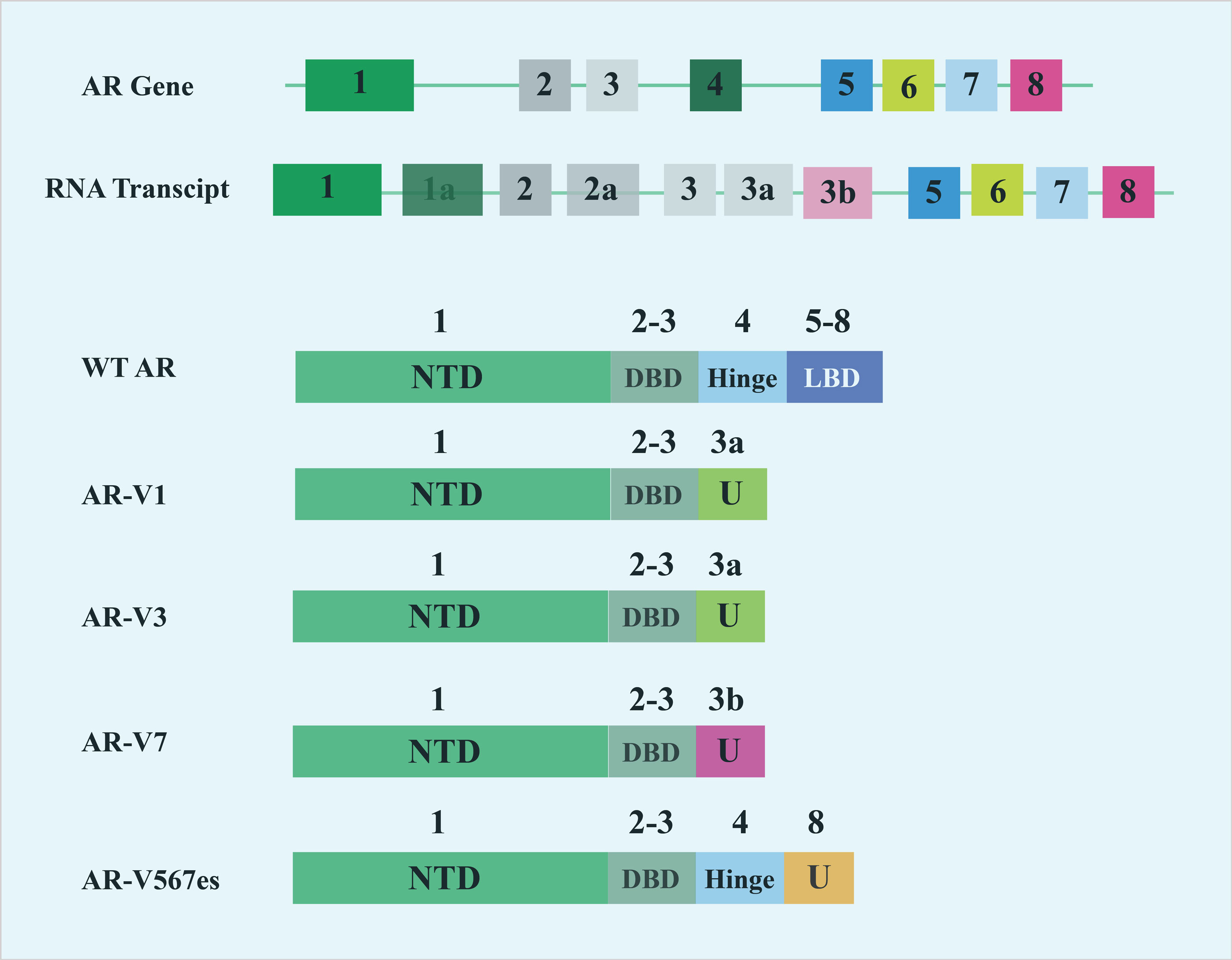 Figure 2. The process of transforming AR family genes into proteins in tumor cells.
Figure 2. The process of transforming AR family genes into proteins in tumor cells.
None.
Ethical policy
Non applicable.
Availability of data and materials
All data generated or analysed during this study are included in this publication.
Author contributions
Maham Khan searched academic literature, wrote the draft manuscript, supervised the review writting progress and approved the final manuscript submission.
Competing interests
Authors report no conflict of interest.
Funding
None.
- Siegel RL, Miller KD, Wagle NS, Jemal A: Cancer statistics, 2023. CA Cancer J Clin 2023, 73(1): 17-48.
- Xia C, Dong X, Li H, Cao M, Sun D, He S, Yang F, Yan X, Zhang S, Li N, et al: Cancer statistics in China and United States, 2022: profiles, trends, and determinants. Chin Med J (Engl) 2022, 135(5): 584-590.
- Li Y, Xie N, Gleave ME, Rennie PS, Dong X: AR-v7 protein expression is regulated by protein kinase and phosphatase. Oncotarget 2015, 6(32): 33743-33754.
- Chen Z, Wu D, Thomas-Ahner JM, Lu C, Zhao P, Zhang Q, Geraghty C, Yan PS, Hankey W, Sunkel B, et al: Diverse AR-V7 cistromes in castration-resistant prostate cancer are governed by HoxB13. Proc Natl Acad Sci-USA 2018, 115(26): 6810-6815.
- Prekovic S, van den Broeck T, Linder S, van Royen ME, Houtsmuller AB, Handle F, Joniau S, Zwart W, Claessens F: Molecular underpinnings of enzalutamide resistance. Endocr Relat Cancer 2018, 25(11): R545-R557.
- Vander Ark A, Cao J, Li X: Mechanisms and Approaches for Overcoming Enzalutamide Resistance in Prostate Cancer. Front Oncol 2018, 8(10): 180-192.
- Linder S, van der Poel HG, Bergman AM, Zwart W, Prekovic S: Enzalutamide therapy for advanced prostate cancer: efficacy, resistance and beyond. Endocr Relat Cancer 2018, 26(1): R31-R52.
- Hamid AR, Pfeiffer MJ, Verhaegh GW, Schaafsma E, Brandt A, Sweep FC, Sedelaar JP, Schalken JA: Aldo-keto reductase family 1 member C3 (AKR1C3) is a biomarker and therapeutic target for castration-resistant prostate cancer. Mol Med 2013, 18(1): 1449-1455.
- Jernberg E, Thysell E, Bovinder Ylitalo E, Rudolfsson S, Crnalic S, Widmark A, Bergh A, Wikström P: Characterization of prostate cancer bone metastases according to expression levels of steroidogenic enzymes and androgen receptor splice variants. PLoS One 2013, 8(11): e77407.
- Penning TM, Jonnalagadda S, Trippier PC, Rižner TL: Aldo-Keto Reductases and Cancer Drug Resistance. Pharmacol Rev 2021, 73(3): 1150-1171.
- Sun SQ, Gu X, Gao XS, Li Y, Yu H, Xiong W, Yu H, Wang W, Li Y, Teng Y, et al: Overexpression of AKR1C3 significantly enhances human prostate cancer cells resistance to radiation. Oncotarget 2020, 11(17): 1575.
- Knudsen KE, Penning TM: Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab 2010, 21(5): 315-324.
- Yepuru M, Wu Z, Kulkarni A, Yin F, Barrett CM, Kim J, Steiner MS, Miller DD, Dalton JT, Narayanan R: Steroidogenic enzyme AKR1C3 is a novel androgen receptor-selective coactivator that promotes prostate cancer growth. Clin Cancer Res 2013, 19(20): 5613-5625.
- Wang B, Gu Y, Hui K, Huang J, Xu S, Wu S, Li L, Fan J, Wang X, Hsieh JT, et al: AKR1C3, a crucial androgenic enzyme in prostate cancer, promotes epithelial-mesenchymal transition and metastasis through activating ERK signaling. Urol Oncol 2018, 36(10): 472.e11-472.e20.
- Yang JC, Xu P, Ning S, Wasielewski LJ, Adomat H, Hwang SH, Morisseau C, Gleave M, Corey E, Gao AC, et al: Novel inhibition of AKR1C3 and androgen receptor axis by PTUPB synergizes enzalutamide treatment in advanced prostate cancer. Oncogene 2023, 42(9): 693-707.
- Tamae D, Mostaghel E, Montgomery B, Nelson PS, Balk SP, Kantoff PW, Taplin ME, Penning TM: The DHEA-sulfate depot following P450c17 inhibition supports the case for AKR1C3 inhibition in high risk localized and advanced castration resistant prostate cancer. Chem Biol Interact 2015, 234: 332-338.
- Adeniji AO, Chen M, Penning TM: AKR1C3 as a target in castrate resistant prostate cancer. J Steroid Biochem Mol Biol 2013, 137: 136-149.
- Zhu S, Ni Y, Wang Z, Zhang X, Zhang Y, Zhao F, Dai J, Wang Z, Zhu X, Chen J, et al: Plasma Exosomal AKR1C3 mRNA Expression Is a Predictive and Prognostic Biomarker in Patients with Metastatic Castration-Resistant Prostate Cancer. Oncologist 2022, 27(11): e870-e877.
- Detlefsen AJ, Mesaros CA, Duan L, Penning TM: AKR1C3 Converts Castrate and Post-Abiraterone DHEA-S into Testosterone to Stimulate Growth of Prostate Cancer Cells via 5-Androstene-3β,17β-Diol. Cancer Res Commun 2023, 3(9): 1888-1898.
- Ning S, Armstrong CM, Xing E, Leslie AR, Gao RY, Sharifi M, Schaaf ZA, Lou W, Han X, Xu DH, et al: LX1 Targets Androgen Receptor Variants and AKR1C3 to overcome Therapy Resistance in Advanced Prostate Cancer. Cancer Res 2024, 2(21): 80-87.
- Luo J, Attard G, Balk SP, Bevan C, Burnstein K, Cato L, Cherkasov A, De Bono JS, Dong Y, Gao AC, et al: Role of Androgen Receptor Variants in Prostate Cancer: Report from the 2017 Mission Androgen Receptor Variants Meeting. Eur Urol 2018, 73(5): 715-723.
- Su CT, Nizialek E, Berchuck JE, Vlachostergios PJ, Ashkar R, Sokolova A, Barata PC, Aggarwal RR, McKay RR, Agarwal N, et al: Differential responses to taxanes and PARP inhibitors in ATM- versus BRCA2-mutated metastatic castrate-resistant prostate cancer. Prostate 2023, 83(3): 227-236.
- Bryce AH, Antonarakis ES: Androgen receptor splice variant 7 in castration-resistant prostate cancer: Clinical considerations. Int J Urol 2016, 23(8): 646-653.
- Gurioli G, Conteduca V, Brighi N, Scarpi E, Basso U, Fornarini G, Mosca A, Nicodemo M, Banna GL, Lolli C, et al: Circulating tumor cell gene expression and plasma AR gene copy number as biomarkers for castration-resistant prostate cancer patients treated with cabazitaxel. BMC Med 2022, 20(1): 48-65.
- Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et al: AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014, 371(11): 1028-1038.
- Zheng Z, Li J, Liu Y, Shi Z, Xuan Z, Yang K, Xu C, Bai Y, Fu M, Xiao Q, et al: The Crucial Role of AR-V7 in Enzalutamide-Resistance of Castration-Resistant Prostate Cancer. Cancers (Basel) 2022, 14(19): 4877.
- Cao S, Zhan Y, Dong Y: Emerging data on androgen receptor splice variants in prostate cancer. Endocr Relat Cancer 2016, 23(12): T199-T210.
- Shah K, Gagliano T, Garland L, O'Hanlon T, Bortolotti D, Gentili V, Rizzo R, Giamas G, Dean M: Androgen receptor signaling regulates the transcriptome of prostate cancer cells by modulating global alternative splicing. Oncogene 2020, 39(39): 6172-6189.
- Fan L, Zhang F, Xu S, Cui X, Hussain A, Fazli L, Gleave M, Dong X, Qi J: Histone demethylase JMJD1A promotes alternative splicing of AR variant 7 (AR-V7) in prostate cancer cells. Proc Natl Acad Sci-USA 2018, 115(20): E4584-E4593.
- Chen Z, Wu D, Thomas-Ahner JM, Lu C, Zhao P, Zhang Q, Geraghty C, Yan PS, Hankey W, Sunkel B, et al: Diverse AR-V7 cistromes in castration-resistant prostate cancer are governed by HoxB13. Proc Natl Acad Sci-USA 2018, 115(26): 6810-6815.
- Jones D, Wade M, Nakjang S, Chaytor L, Grey J, Robson CN, Gaughan L: FOXA1 regulates androgen receptor variant activity in models of castrate-resistant prostate cancer. Oncotarget 2015, 6(30): 29782-29794.
- Liu C, Lou W, Yang JC, Liu L, Armstrong CM, Lombard AP, Zhao R, Noel ODV, Tepper CG, Chen HW, et al: Proteostasis by STUB1/HSP70 complex controls sensitivity to androgen receptor targeted therapy in advanced prostate cancer. Nat Commun 2018, 9(1): 4700.
- Li Y, Xie N, Gleave ME, Rennie PS, Dong X: AR-v7 protein expression is regulated by protein kinase and phosphatase. Oncotarget 2015, 6(32): 33743-33754.
- Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, Tien AH, Yang YC, Tavakoli I, Haile S, Watt K, et al: An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest 2013, 123(7): 2948-2960.
- Henzler C, Li Y, Yang R, McBride T, Ho Y, Sprenger C, Liu G, Coleman I, Lakely B, Li R, et al: Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat Commun 2016, 29(7): 13668.
- Paschalis A, Sharp A, Welti JC, Neeb A, Raj GV, Luo J, Plymate SR, de Bono JS: Alternative splicing in prostate cancer. Nat Rev Clin Oncol 2018, 15(11): 663-675.
- Li Y, Alsagabi M, Fan D, Bova GS, Tewfik AH, Dehm SM: Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res 2011, 71(6): 2108-2117.
- Li Y, Hwang TH, Oseth LA, Hauge A, Vessella RL, Schmechel SC, Hirsch B, Beckman KB, Silverstein KA, Dehm SM, et al: AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene 2012, 31(45): 4759-4767.
- Sciarra A, Maggi M, Salciccia S, Nicolai A, Tortorella E, Giantulli S, Magliocca FM, Silvestri I, Taglieri L, Cattarino S, et al: Tissue Expression of Androgen Receptor Splice Variant 7 at Radical Prostatectomy Predicts Risk of Progression in Untreated Nonmetastatic Prostate Cancer. Oncology 2021, 99(4): 251-255.
- Nilsen TW, Graveley BR: Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463(7280): 457-463.
- Dreyfuss G, Kim VN, Kataoka N: Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol 2002, 3(3): 195-205.
- Lee Y, Rio DC: Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu Rev Biochem 2015, 84: 291-323.
- Carabet LA, Leblanc E, Lallous N, Morin H, Ghaidi F, Lee J, Rennie PS, Cherkasov A: Computer-Aided Discovery of Small Molecules Targeting the RNA Splicing Activity of hnRNP A1 in Castration-Resistant Prostate Cancer. Molecules 2019, 24(4): 763.
- Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Chen H, Kong X, Melamed J, Tepper CG, et al: A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res 2009, 69(6): 2305-2313.
- Wang B, Wu S, Fang Y, Sun G, He D, Hsieh JT, Wang X, Zeng H, Wu K: The AKR1C3/AR-V7 complex maintains CRPC tumour growth by repressing B4GALT1 expression. J Cell Mol Med 2020, 24(20): 12032-12043.
- Fan L, Peng G, Hussain A, Fazli L, Guns E, Gleave M, Qi J: The Steroidogenic Enzyme AKR1C3 Regulates Stability of the Ubiquitin Ligase Siah2 in Prostate Cancer Cells. J Biol Chem 2015, 290(34): 20865-20879.
- Liu C, Yang JC, Armstrong CM, Lou W, Liu L, Qiu X, Zou B, Lombard AP, D'Abronzo LS, Evans CP, et al: AKR1C3 Promotes AR-V7 Protein Stabilization and Confers Resistance to AR-Targeted Therapies in Advanced Prostate Cancer. Mol Cancer Ther 2019, 18(10): 1875-1886.
- Liu Y, He S, Chen Y, Liu Y, Feng F, Liu W, Guo Q, Zhao L, Sun H: Overview of AKR1C3: Inhibitor Achievements and Disease Insights. J Med Chem 2020, 63(20): 11305-11329.
- Tian Y, Zhao L, Wang Y, Zhang H, Xu D, Zhao X, Li Y, Li J: Berberine inhibits androgen synthesis by interaction with aldo-keto reductase 1C3 in 22Rv1 prostate cancer cells. Asian J Androl 2016, 18(4): 607-612.
- Penning TM: Aldo-Keto Reductase (AKR) 1C3 inhibitors: a patent review. Expert Opin Ther Pat 2017, 27(12): 1329-1340.
- Penning TM: AKR1C3 (type 5 17β-hydroxysteroid dehydrogenase/prostaglandin F synthase): Roles in malignancy and endocrine disorders. Mol Cell Endocrinol 2019, 489: 82-91.
- Kikuchi A, Furutani T, Azami H, Watanabe K, Niimi T, Kamiyama Y, Kuromitsu S, Baskin-Bey E, Heeringa M, Ouatas T, et al: In vitro and in vivo characterisation of ASP9521: a novel, selective, orally bioavailable inhibitor of 17β-hydroxysteroid dehydrogenase type 5 (17βHSD5; AKR1C3). Invest New Drugs 2014, 32(5): 860-870.
- Loriot Y, Fizazi K, Jones RJ, Van den Brande J, Molife RL, Omlin A, James ND, Baskin-Bey E, Heeringa M, Baron B, et al: Safety, tolerability and anti-tumour activity of the androgen biosynthesis inhibitor ASP9521 in patients with metastatic castration-resistant prostate cancer: multi-centre phase I/II study. Invest New Drugs 2014, 32(5): 995-1004.
- Hilpert J, Groettrup-Wolfers E, Kosturski H, Bennett L, Barnes CLK, Gude K, Gashaw I, Reif S, Steger-Hartmann T, Scheerans C, et al: Hepatotoxicity of AKR1C3 Inhibitor BAY1128688: Findings from an Early Terminated Phase IIa Trial for the Treatment of Endometriosis. Drugs R D 2023, 23(3): 221-237.
- Battista C, Shoda LKM, Watkins PB, Groettrup-Wolfers E, Rottmann A, Raschke M, Generaux GT: Quantitative Systems Toxicology Identifies Independent Mechanisms for Hepatotoxicity and Bilirubin Elevations Due to AKR1C3 Inhibitor BAY1128688. Clin Pharmacol Ther 2023, 114(5): 1023-1032.
- Siegel RL, Miller KD, Wagle NS, Jemal A: Cancer statistics, 2023. CA Cancer J Clin 2023, 73(1): 17-48.
- Xia C, Dong X, Li H, Cao M, Sun D, He S, Yang F, Yan X, Zhang S, Li N, et al: Cancer statistics in China and United States, 2022: profiles, trends, and determinants. Chin Med J (Engl) 2022, 135(5): 584-590.
- Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, et al: Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res 2009, 69(1): 16-22.
- Ahrens-Fath I, Politz O, Geserick C, Haendler B: Androgen receptor function is modulated by the tissue-specific AR45 variant. FEBS J 2005, 272(1): 74-84.
- Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ: Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res 2008, 68(13): 5469-5477.
- Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et al: AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014, 371(11): 1028-1038.
- Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Nakazawa M, Nadal R, Paller CJ, Denmeade SR, Carducci MA, et al: Androgen Receptor Splice Variant 7 and Efficacy of Taxane Chemotherapy in Patients With Metastatic Castration-Resistant Prostate Cancer. JAMA Oncol 2015, (5): 582-591.
- Ma Y, Luk A, Young FP, Lynch D, Chua W, Balakrishnar B, de Souza P, Becker TM: Droplet Digital PCR Based Androgen Receptor Variant 7 (AR-V7) Detection from Prostate Cancer Patient Blood Biopsies. Int J Mol Sci 2016, 17(8): 1264.
- Hodara E, Morrison G, Cunha A, Zainfeld D, Xu T, Xu Y, Dempsey PW, Pagano PC, Bischoff F, Khurana A, et al: Multiparametric liquid biopsy analysis in metastatic prostate cancer. JCI Insight 2019, 4(5): e125529.
- Li H, Wang Z, Tang K, Zhou H, Liu H, Yan L, Guan W, Chen K, Xu H, Ye Z: Prognostic Value of Androgen Receptor Splice Variant 7 in the Treatment of Castration-resistant Prostate Cancer with Next generation Androgen Receptor Signal Inhibition: A Systematic Review and Meta-analysis. Eur Urol Focus 2018, 4(4): 529-539.
- Qu Y, Dai B, Ye D, Kong Y, Chang K, Jia Z, Yang X, Zhang H, Zhu Y, Shi G: Constitutively active AR-V7 plays an essential role in the development and progression of castration-resistant prostate cancer. Sci Rep 2015, 5: 7654.
- Welti J, Rodrigues DN, Sharp A, Sun S, Lorente D, Riisnaes R, Figueiredo I, Zafeiriou Z, Rescigno P, de Bono JS, et al: Analytical Validation and Clinical Qualification of a New Immunohistochemical Assay for Androgen Receptor Splice Variant-7 Protein Expression in Metastatic Castration-resistant Prostate Cancer. Eur Urol 2016, 70(4): 599-608.
Annals of urologic oncology
p-ISSN: 2617-7765, e-ISSN: 2617-7773
 Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.