
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
Roles of Renin-Angiotensin System in the Regulation of Prostate Cancer Bone Metastasis: A Critical Review
Manh Tien Tran1
1Department of Dental Pharmacology, Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama University, Okayama 700-8525, Japan. Correspondence: Manh Tien Tran (Department of Dental Pharmacology, Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama University, Okayama 700-8525, Japan; Email: trantienmanh1508@gmail.com).
Annals of Urologic Oncology 2021, 4(1): 31-41. https://doi.org/10.32948/auo.2021.10.20
Received: 31 Aug 2021 | Accepted: 07 Oct 2021 | Published online: 15 Oct 2021 
Mestastatic prostate cancer cells (MPCCs) frequently metastasize to bone, which is a “favorite soil” for colonization and proliferation of MPCCs. Prostate cancer bone mestastasis is tightly associated with tumor-induced bone lesions, most commonly caused from (1) the etiological imbalance between osteoblastic bone formation and osteoclastic bone resorption and from (2) the anti-tumor immune response. Therefore, understanding of prostate cancer biology and prostate cancer bone metastasis has led to the establishment of drug development programs for treatment of the patients with bone metastasis. The renin-angiotensin system (RAS) controls systemic body fluid circulation; nonetheless, the existence of a local RAS in tumors has been reported. Importantly, the local RAS has recently emerged as a potential regulator of tumorigenesis and cancer metastasis. This review summarizes and dissects the critical roles of the local RAS in promoting (1) progression of metastatic prostate cancer, and (2) development and progression of PCa bone metastasis, thereby providing multiple solutions for the potential therapeutic intervention.
Key words RAS, metastatic PCa, PCa bone metastasis, bone microenvironment
Additionally, the activated AT1R could also be associated with the soluble and receptor tyrosine kinases (RTKs), the mitogen-activated protein kinases (MAPKs) such as extracellular-regulated kinases (ERK1/2, p38 MAPK and Jun N-terminal kinase) [10, 11], the JAK-STAT pathway [12]. Moreover, it also mediates the generation of reactive oxygen species (ROS) [13] and the modulation of various ion channels [14-16]. The mechanisms underlying the AT1R-triggered regulation of tyrosine kinase-related pathways are thought to initiate from (1) the de novo synthesis of cytokine and growth factor ligands, and/or (2) the binding of the AT1R with the upstream activators and scaffolds [17], (3) and/or the transactivation of growth factor receptors such as the epidermal growth factor receptor (EGFR) [18]. EGFR transactivation involves the capacity of GPCRs to activate metalloproteinases (MMPs) that cleave cell surface precursors of the EGF ligands, yielding the shed mature ligand that subsequently binds and activates the EGFR to pioneer various cellular signals [19], which are crucial for Ang II/AT1R-mediated hypertrophy and/or proliferation of cardiac, vascular and renal cells. The signaling pathways regulated by the other RAS receptors (AT2R, MASR, IRAP, renin receptor, ACE and ACE2) are poorly understood. However, several of the published reports have elucidated that AT1R, AT2R, MASR [20] and other members of GPCR family [21] could form dimerization, which declined the efficacy and the potency of ligand binding; however, the physiogical effects of the dimerization of these receptors to the normal and disease states have been controversial. As the RAS plays a central role in (1) the elevation of blood pressure, (2) the maintainance of fluid homeostasis, the dysregulation of RAS has been tightly linked to hypertension and cardiovascular diseases.
Bone is a metaboligcally active tissue continuously remodeled throughout life. Bone remodeling is a process in which the old/damaged bone is removed and replaced with newly synthesized bone to maintain bone strength and mineral homeostasis [22]. Bone homeostasis is tightly controlled by (1) osteoblasts (OBs), which are responsible for synthesizing bone matrix proteins and promoting bone deposition and mineralization, and (2) osteoclasts (OCs), which are responsible for removing both the mineral and the organic matrix of bone [23]. Mechanical forces, proinflammatory cytokines, growth factors and hormones trigger an imbalance between osteoblastic and osteoclastic activities, which are one of the major causes of the bone-related diseases [24] such as postmenopausal osteoporosis, hyperparathyroidism, rheumatoid arthritis and osteopetrosis. In addition to the lungs and liver, bone is one of the most frequent sites of metastases. Prostate and breast cancers are responsible for the majority of bone metastases [25]. Patients with bone metastasis experience a very miserable quality of life as a consequence of severe pain, spinal cord compression, fractures, bone marrow aplasia and hypercalcermia, the latter being a principal cause of death. In fact, hypercalcemia can induce gastrointestinal dysfunctions, as well as constipation, polyuria and fatigue. In the advanced stage, it is the major cause of renal failure and cardiac arrhythmias. All these symptoms are usually classified as skeletal-related events (SREs) [26].
PCa arising in the prostate gland is the most common type of malignancy that has an tendency to develop in older men, aged 50 and over [27]. When PCa cells metastasize, they most frequently spreads to bone. Advanced-stage PCa is characterized by tropism to bone with skeletal involvement present in approximately 90% of patients with metastatic disease. Metastatic PCa is characterized by a period of responsiveness to castration, and metastatic, castration-resistant prostate cancer (mCRPC) frequently leads to skeletal complications such as ineffective haematopoiesis, pain and skeletal related events (SREs) [28]. The term SRE is used to encompass common complications of bone metastases such as pathologic fracture secondary to a bone metastasis, spinal cord compression or the need for surgery or radiotherapy to bone [28].
Recent studies have shown several components of the RAS (renin, ACE, and Ang II receptors) were expressed in the bone marrow microenvironment, and play an important role in bone metabolism [29, 30]. Besides, the local bone RAS might participate in regulating the pathogenesis and progression of some metabolic bone diseases via two distinctive ways: (1) an imbalance in bone remodeling, characterized by the inhibition of osteoblastic activity, or (2) the stimulation of osteoclastic activity [31, 32]. Additionally, accumulating clues obtained from in vitro, animal and clinical studies, have substantiated a frequent dysregulation of RAS in metastatic tumor cells, in correlation with poor diagnosis in patients. Antagonism/blockade of the RAS by the specific antagonists/blockers suppress tumor growth, angiogenesis and metastasis in a broad range of experimental models of malignancy, and retrospective studies in humans, providing clues that a long-term use of angiotensin-converting enzyme (ACE) inhibitors may protect against cancer. In this article, I review and summarize the current knowledge on the specific roles of the RAS (1) in regulating the metastatic PCa progression in the primary (prostate) gland, and (2) in maintaining the functional interactions amongst the metastatic PCa cells, bone cells and immune cells.
PCa is the majority of malignancies arising from the peripheral portion of the gland surrounding the prostatic urethra just below the bladder [33]. Two disorders commonly affecting the prostate are (1) non-cancerous enlargement of the prostate, known as the benign prostatic hyperplasia (BPH), and (2) Pca. The initial feature of PCa bone metastasis is that PCa cells not only shed many of their epithelial characteristics, also known as mesenchymal-epithelial transition (EMT), but crucially acquire the metastatic capacities such as motility and invasiveness, to detact from the prostate epithelium.
Many published reports have proven that a cancer incidence rate has been significantly reduced in cancer patients undergoing a long-term treatment with RAS-specific inhibitors. Perindopril-triggered blockade of ACE activity suppressed the VEGF-mediated tumor development and neovascularization in murine hepatocellular carcinoma cells [34]. Besides, Mamoru Fujita et al, found that blockade of ACE activity by a non-peptide AT1R antagonist (TCV-16) resulted in the (1) alleviation of the tumor angiogenesis and tumor growth in mice subcutaneously injected with sarcoma and fibrosarcoma cell lines [35], and (2) the impairment of the metastatic ability of Lewis lung carcinoma (3LL) cells, injected intravenously [35]. In addition, olmesartan-induced blockade of ACE and AT1R activities suppressed the growth of subcutaneous tumors generated by the co-injection of pancreatic cancer cells with pancreatic stellate cells (PSCs) [36] whereas losartan-induced blockade of AT1R activity inhibited angiogenesis and tumor growth in mice subcutaneously injected with C6 rat glioma [37], which is the most frequent brain tumor [38]. Moreover, directly acting on AT1R, Ang II was demonstrated to be crucial for promoting (1) the invasive ability and VEGF seceretion in SKOV-3 ovarian cancer cells [39], (2) promote the expression of MMP2 and MMP9 in MKN-28 cells. However, the stimulatory effects of Ang II on microvessel and VEGF expression could be abrogated by AT1R antagonist [40, 41]. Additionally, Ang (1-7) has an anti-metastatic action on lung adenocarcinoma cells [42, 43]. Ultimately, AT2R overpexression triggered (1) the cell cycle arrest at S phase, and (2) apoptosis via an extrinsic cell death signaling pathway in a manner dependent upon the activation of p38 MAPK, caspase-8 and caspase-3 in the PCa cell line, Du145 [44]. Besides, Ang II abundantly expressing in non-basal epithelial cells in prostate, directly stimulated the growth of prostate cancer cell lines, LNCaP and Du145, via AT1Rs [45]. Ang II-mediated AT2R stimulation diminished EGF-induced ERK2 phosphorylation in both LNCaP and PC3 cell lines. ERK2 activation is regulated by two independent pathways comprising (1) Ras/Raf-dependent pathway involving transactivation of the EGFRs mediated by heparin binding epidermal growth factor (EGF)-like growth factor (HB-EGF) [46], and (2) mediated by a PKC-dependent pathway [47]. The intracellular mechanisms of how AT2R could inhibit ERK2 phosphoryation is poorly understood; however, katarina Bedecs et al. once reported an important role of Ang II in rapidly activating the protein tyrosine phosphatase-1 (SHP-1), thereby terminating the cytokine and/or growth-factor receptor-mediated signaling pathways in the neuroblastoma cell line, N1E-115 and the the Chinese hamster ovary cell line expressing recombinant human AT2R [48]. Of greater potential significance, Ang II played a pivotal role in regulating the cell proliferation in LNCaP cells expressing both functional AT1R and AT2R whereas it served as an negative regulator of cell growth in PC3 cells expressing non-functional AT1R, evidently indicating that the autocrine of Ang II on cell growth can vary dependently upon the functional state of both AT1R and AT2R [45]. Also, Ang II could act on the regulation of the signaling transducers such as MAPK and STAT3. Toshiaki Ishizuka et al. revealed (1) that stimulation of mouse induced pluripotent stem (iPS) cells with AT1R enhanced LIF-induced DNA synthesis via mediating the JAK/STAT3 signaling pathway, and (2) that AT1R stimulation enhanced mouse iPS cell differentiation into the mesodermal progenitor cells via p38 MAPK activation [49]. Intriguingly, Kotha et al. had reported that resveratrol induced apoptosis in human PCa (Du145) cells by blocking STAT3 signaling pathway [50] before Mee-huyn lee et al. revealed the suppressive effects of resveratrol on IL-6-induced androgen receptor (AR) transcriptional activity in LNCaP PCa cells partly via the inhibition of STAT3 reporter gene activity [51].
In addition, the elevated levels of EGF factor and its specific receptor, EGFR, were confirmed in PCa cells as well as the clinical specimens, suggesting that EGFR might have involved in the regulation of the prostate cancer progression. For instance, caveolin-1 (Cav-1), overexpressed in PCa and involved in the development and the progression of PCa, was involved in the regulation of the cellular signaling proteins within caveolae where it interacts with the receptor tyrosine kinases (RTKs), serine/threonine kinases (STKs), phospholipases (PLs), G protein-coupled receptors (GPCRs), Src family kinases (SFKs), and AT1Rs. Cav-1 expression was increased, following Ang II treatment; nonetheless, candesartan (AT1R blocker)-induced blockade of AT1R strongly reduced the Cav-1 expression, subsequently inhibiting the growth of the AngII-treated metastatic prostate adenocarcinoma PC3 and Du145 cells [52, 53]. Although the IP3K/Akt pathway is the primary inducer of EMT, the Wnt/β-catenin, Notch, Ras, integrin-linked kinase, and integrin signaling pathway are also involved [54-57]. Collectively, AT1-R and/or AT2-R blockers can be used as the potent inhibitors of MAPK, STAT3, IP3K/Akt pathways, thereby alleviating the PCa progression; accordingly, these blockers would be the potent anticancer drugs for treating patients with PCa (table 1).
1. Functional Interplay amongst metastatic PCa cells and bone cells
Osteoblastic bone formation is mediated by synthesizing the extracellular matrix (ECM) mostly including the dense lammelar type I collagen [58]. ECM, known as the osteoid, is sequentially deposited and mineralized via the accumulation of calcium phosphate as hydroxyapatite [59]. Multiple transcription factors such as RUNX2 and OSX and major developmental signals such as WNT signaling are reported to regulate OB differentiation and function [60]. Reversely, bone resorption is regulated by OCs, the large, multinucleated cells formed by the fusion of precursors from the monocyte-derived macrophage lineage. Functionally, OCs can dissolve minerals and digest bone matrix by secreting hydrochloric acid and proteolytic enzymes [61]. Similar to OBs, multiple factors are identified to regulate OC differentiation, function and survival, consisting of macrophage colony-stimulating factor (M-CSF), receptor activator of nuclear factor κB ligand (RANKL), cytokines (for example, IL-1) and αVβ3 integrin [62]. RANKL, a transmembrane protein, is expressed by OBs, stromal cells, cancer cells and T lymphocytes. The proteolytic cleavage of RANKL generates its soluble form, subsequently binding and activating its RANK receptors that are abundantly expressed on cell surface of the osteoclast precursors [63]. In contrast, osteoprotegerin (OPG), also known as tumor necrosis factor receptor (TNFR) superfamily member 11B secreted by OBs [64], functions as a decoy receptor for RANKL, abolishes RANK-RANKL signaling. RANK-RANKL-OPG signaling is of critical importance to the maintenance of normal bone.
RAS plays an important role in bone metabolism, especially osteoporosis, a bone disease characterized by losses of bone mineral density and bone mass [65]. Previous studies have shown the expression of renin in bone marrow cells, and ACE in OBs and OCs [66]. In bone tissue, Ang II produced locally by the vascular endothelial cells exerts its action by binding to AT1R and AT2R, both of which are expressed in OBs. Etiologically, it was revealed that Ang II could stimulate the differentiation and the activity of OCs in vivo and in vitro and aggravate the loss of bone minerals in rats with osteoporosis induced by estrogen deficiency; furthermore, the AT1R knockout mice showed high bone mass. For instance, sa-sa Gu et al. reported that the local bone RAS was involved in age-related osteoporosis of aging mice [30], bone deteriorations of mice with either obstructive nephropathy [67] or type 1 diabetes [68], indicating that the local bone RAS displays a pivotal role in the regulation of bone tissue homeostasis. Particularly, the interaction of Ang II with AT1R might be involved in the regulation of the OB-rich populations of cells through AT1R [32, 69]. Besides, both in vitro and in vivo studies have elucidated that Ang II was responsible for OC activation via promoting the OB-induced secretion of cytokines and/or RANK ligands [70]. The Ang II-mediated secretion of the matrix metalloproteinase (MMP)-3 and -13 via the AT1 receptor into the ECM in osteoblastic ROS17/2.8 cells accelerated bone resorption [71]. Notably, Ang II neither affected OC-induced bone resorption in the isolated OCs nor potentiate the RANKL-induced OC formation; however, it could stimulate RANKL-induced osteoclastogenesis in the OB and OC co-cultures [31, 72]. Moreover, Ang II-triggered activation of AT1R markedly inhibited OB differentiation and mineralization and alleviated the mineralized nodule formation of rat calvarial osteoblastic cells [31, 73].
PCa bone metastasis is commonly associated with osteoblastic lesions, generally arising from an imbalance between bone forming OBs and bone-resorbing OCs in prostate cancer. While bone mestastasis from breast and lung cancers frequently induce osteolytic or the bone-lysing lesions, PCa bone metastasis uniquely triggers bone formation, suggesting OBs enables PCa cells to metastasize to bone. More detailed, PCa bone metastasis is thought to originate from the functional interactions between MPCCs and the bone microenvironment, in general, bone cells, in particular. For instance, Li et al. showed that OBs promoted the growth of PCa cells in a co-culture model (androgen-independent C4-2 PCa cells and osteoblastic cells) [74]; besides, Gleave el al. demonstrated that co-inoculation of the athymic mice with human PCa cells (LNCaP) and with the human bone fibroblasts promoted the formation of LNCaP cell-derived tumor [75]. Later, Sung et al. reported that co-cultrure of human bone stromal cells with human PCa cells under 3D cultured condition enhanced tumor xenografts in mice [76]. Besides, hydrochloride-triggered blockade of bone morphogenetic protein 4 (BMP-4) receptor activation in OBs diminished the growth of PCa-18b tumors in mice [77], clarifying that BMP4-mediated osteoclastogenesis promotes the development and the progression of PCa bone metastasis in mice. Co-culturing human bone stromal cells with PCa cells in vitro, triggered the elevation of expression levels of ECM (versican and tenascin) and chemokines (BDFN, CCL5, CXCL5 and CXCL16) genes [76]; furthermore, using the bone metastasis exnograft models of human osteoinductive PCa cells (VCaP and C4-2B) to generate the osteoblastic bone metastasis-associated stromal transcriptiome (OB-MDST), Berna C. O¨ zdemir et al. identified the up-regulation of PTN, EPHA3 and FSCN1 in mouse stromal cells [78]. However, how these stromal secreted factors provide a support for PCa bone metastasis needs to be further investigated.
OC activation has been associated with the development of PCa bone metastasis [79, 80]. Critically, it is worthy noting that OC differentiation might be originated from the Cbfa1/Runx2-induced secretion of RANKL, following Ang II-mediated OB activation. Cbfa1/Runx2 is an essential transcription factor for the differentiation of OBs from the mesenchymal precursors [81]. Ang II increased the [cAMP]i either through interacting with AT1R in cultured OBs [31] or through facilitating the interaction between activated Gs and adenylyl cyclase (AC) in cultured rat vascular smooth muscle cells [82]. Elevation of [cAMP]i results in the activation of the downstream signaling cascades, which enhance the RANKL secretion as well as down-regulate the transcriptional function of Cbfa1/Runx2, leading to the alleviation of OB activation and bone formation [70, 83]. Mature OCs subsequently adhere to the bone surface through αvβ3 integrin, form ruffled borders, and secrete acid to solubilize calcium phosphate crystals as well as secret the collagenases and proteinases such as tartrate-resistant acid phosphatase (TRAP), matrix metallopeptidase 9 (MMP9), and cathepsin K (CTSK) that demineralize and degrade extracellular proteins such as type I collagen. The release of bone resorption-derived Ca2+ ions triggers activation of Ca2+-sensing receptor (CaR), a G protein-coupled receptors (GPCRs), on PM of OBs [84], OCs [85] and especially MPCCs [86, 87], subsequently stimulating the secretion of parathyroid hormone-related protein (PTHrP) from MPCCs (PC-3 cell line) [86, 87]. PTHrP secreted by PCa cells could then activate OCs and potentially contribute towards the skeletal invasiveness, bone pain, and/or pathological fractures. Additionally, the transforming growth factor beta, TGFβ, a bone resorption-derived factor and a key mediator of bone metastasis, stimulates PTHrP expression in OBs [88] and tumor cells [89], thereby promoting osteolysis. Specifically, TGFβ-mediated signaling pathway activating a couple of vital intracellular cascades such as MAPK, PI3K/Akt, and Rho-like GTPase signaling cascades, critically acts as a driver of tumor progression and bone metastasis. Besides, bone marrow produces the factors, such as CXCL12, with a chemotactic role on cancer cells, which, on the other hand, express the chemokine receptors, CXCR4 and CXCR7 [90], thereby facilitating the establishment of “fertile soil” that promotes the tumor cells to adhere to bone matrix and thrive in bone. Moreover, cancer cells, in turn, secrete prostaglandins, parathyroid hormone (PTH), PTHrP, activated vitamin D, IL-6 and TNF that may lead to an elevation of RANKL expression on OBs and bone marrow stromal cells, resultantly stimulating the formation, survival and activity of OCs, thereby promoting osteolytic metastases. Notch-Jagged interactions in the bone marrow suggest direct activation of osteolysis by cancer cells through this unique interaction. In particular, Jagged1, which is a downstream mediator of the prometastatic TGFβ, promotes tumor growth through stimulating IL-6 production from OBs, and directly it activates OC differentiation (Figure 1). Moreover, Jagged1 is overexpressed by the mestastatic PCa cells with bone metastasis [91], whereas its Notch receptor is frequently expressed by progenitors and mature cells in the bone marrow [92]. Interestingly, metastatic PCa cells respond to growth factor stimulation via the activation of various osteoblastic transcription factors. This would suggest that bone lesions may also occur through the differentiation of the cancer cells towards an osteoblastic bone-forming phenotype, which is a phenomenon that has been observed in PCa bone metastasis.
2. Functional interplay amongst metastatic PCa cells and immune cells
Bone, immune and hematopoietic systems are tightly linked since bone cells and hematopoietic cells are in deep physical contact, are reciprocally regulated, are interconnected in their function, and share several common pathways. Indeed, bone cells express surface molecules regulating the expansion of hematopoietic stem cells (HSCs) from which all cells of the mammalian immune system derive, whereas many immunoregulatory cytokines directly act on bone cells. OBs and OCs both affect the maintenance and the mobilization of HSCs [93]. OBs control the proliferation of hematopoietic progenitors and the differentiation of all stages of B cell development. Indeed, in vitro production of B cell precursors from progenitors required contact with OBs and expression of CXCL12 and IL-7, which was induced by PTH [94]. Besides, OC precursors, T, B and NK cells are derived from the HSCs; accordingly, some of the receptors and ligands participating in regulating the immune processes also modulate the maturation of OC precursors and the bone-resorbing activity of active OCs. RANKL, its receptor RANK, and OPG form a crucial molecular link between the immune system and bone [95]. The membrane RANKL is expressed by stromal cells, OBs and metatastic PCa cells [96, 97]. The soluble RANKL was produced not only by the synovial fibroblasts, T helper 17 (Th17) cells [98, 99], but it is also expressed by B cells and PCa cells while RANK receptors are expressed by dendritic cells (DCs), monocytes, macrophages and PCa cells [100, 101]. Th17 cell- secreted RANKL directly regulates osteoclastogenesis and bone remodeling. Besides, the vesicular RANK secreted from OCs promotes osteoblastogenesis by activating Runx2 via RANK-RANKL interaction [102]. Many immune factors such as interferon-γ (IFN-γ) regulating osteoclastogenesis directly by inhibiting OC maturation [103] or indirectly by activating T cells, followed by the elevation of proosteoclastogenic factors [104].
Another important mediator of the interactions between T cells and bone cells is IL-7, a cytokine produced by stromal cells at the inflammatory sites, with different effects on hematopoietic and immunologic systems [105]. The main function of IL-7 is the control of B and T lymphopoiesis, but it significantly contributes towards the invasiveness of PCa cells by promoting EMT [106], and regulates bone homeostasis [107]. IL-7, produced by T cells, promotes osteoclastogenesis by upregulating T cell-derived cytokines, such as RANKL and TNFα [108]. However, OCs also plays a pivotal role in regulating the T cell function because it present the antigenic peptides to T cells and induce FoxP3 expression in CD8+ T cells [109], which regulate an inappropriate activation of the immune response. The interaction between T cells and OCs are tightly controlled by the reciprocal CD137/CD137L and RANKL/RANK interactions [110]. CD137 (4-1BB), a costimulatory member of the TNF receptor family, facilitates enhanced proliferation, cytokine production, and cytolytic activity of T and NK cells [111]. Ligation of its ligand, CD137L, constitutively expressed in OC precursors, inhibited RANKL-induced OC formation [110]; on the other hand, ligation of RANKL with OPG-Fc, the decoy receptor for RANKL, inhibited both CD4+ and CD8+ T cell proliferation [110].
T and NK cells are fully equipped with RAS components and are potentially capable of producing and delivering AngII to sites of inflammation. Ang II was shown to trigger significant leukocyte rolling, adhesion, and emigration, contributing not only to hypertension, but also to the onset and progression of the vascular damage associated with disease states [112]. Ang II, acting through the AT1R on immune cells, triggers the proliferation of splenic lymphocytes through a calcineurin-dependent pathway, which leads to the possibility of using RAS blockade as an anti-inflammatory and immunosuppressive therapy [113]. The stimulatory function of AngII on leukocyte proliferation seems to be mediated by both types of AngII receptors. The action of the RAS on T cells is mediated through several cytokines, particularly TNFα [114]. Inhibition of ACE diminished TNFα production in T cells [114]. Angiotensin receptor blockers (ARBs) have been shown to suppress lymphocyte proliferation and IFN-γ production, leading to the suppression of the antigen-specific Th1 and Th2 immune responses and suggesting another pathway for T cell activation [115]. In addition, Jon A Weidanz et al. reported that blockade of AT1R inhibited IFN-γ production in the cytotoxic T cells, suggesting that AT1R blockers may have a clinically relevant immunomodulatory role by blocking IFN-γ production in T cells [116].
However, very little is known about the roles of the RAS in mediating the functional interactions between immune cells and the metastatic PCa cells in bone milieu. Therefore, more works would be needed to verify whether the RAS is involved in the regulation of PCa bone metastasis.
3. RAS and mCRPC
The mCRPC, originating from metastatic hormone sensitive PC (mHSPC), is the advanced form not responding to initial treatments such as surgery and hormone therapy while mHSPC still responds to standard hormone treatment that is known as androgen deprivation therapy (ADT). The mCRPC is poorly diagnostic, and commonly associated with significant morbidity, and bone metastases along with the related complications that represent the major cause of death. To date, the mainstay treatment methods applied to patients with mHSPC is mostly based on ADT in combination with docetaxel chemotherapy and oral anti-androgens lead to a significant reduction of the level of circulating testosterone to “castrate level”, subsequently resulting in PCa cell apoptosis. However, PCa cells eventually become resistant to therapy with signs and/or symptoms of progression observable in most patients, in spite of good initial responses.
The recent results of phase III randomized trials showed that blockade of the androgen receptor (AR) through either inhibiting androgen synthesis or directly targeting AR can improve survival for patients with mCRPC by the use of gonadotropin-releasing hormone (GnRH) agents (agonists or antagonists) or orchiectomy [117]. The oncogenic roles of the ARs and AR signalings essential for CRPC development has been intensively investigated in recent years. Prostate-specific antigen (PSA) gene expression is regulated by the AR pathway as the androgen response elements (AREs) are found in the promoter region of the PSA gene. In HSPC, ablation of testicular androgens resulted in alleviating the serum PSA levels, suggesting that AR signaling pathways were down-regulated in a castrate environment [118]. Nevertheless, it was confirmed that PSA levels were increased during disease progression in most patients, suggesting that AR signaling pathways might have been reactivated in a castration-resistant manner. Consequently, more studies will be required to further understand the signaling transduction mechanisms underlying castration resistance, thereby facilitating the development and approval of novel AR-directed drugs such as, for instance, abiraterone acetate plus prednisone (AAP), enzalutamide and taxanes [119]. The patients with mCRPC can be treated with docetaxel or AR-targeted therapies as first-line therapy [120]. However, Vincenza Conteduca et al. has reported that not only the circulating AR aberrations are associated with worse outcome on abiraterone/enzalutamide [121], but also plasma AR gain in docetaxel-treated patients was associated with significantly shorter overall survival (OS) [122]. More importantly, docetaxel can also be associated with toxic effects, comprising fatigue and myelosuppression, and many patients with CRPC are either not healthy sufficient to receive docetaxel or unwilling to receive docetaxel. Despite advances in the treatment of CRPC, patients ultimately progress; a need remains for effective and tolerable treatments, including those who are unsuitable for or decline docetaxel.
Radium-223 dichloride (randium-223), a novel targeted α-emitter, selectively binds to areas of increased metabolic activity in bone metastases. In principle, it emits the high-energy α-particles with ultra-short penetration (<100 μm; 2-10 cell diameters) so as to efficaciously generate cytotoxicity in target areas, whereas reducing the damages to surrounding normal tissue, comprising bone marrow [123]. The phase 3 ALSYMPCA (ALpharadin in SYMptomatic Prostate Cancer patients) trial of radium-223 plus best standard of care versus placebo plus best standard of care in patients with CRPC and symptomatic bone metastases was unique as it is applicable for both groups of patients (1) who had received previous docetaxel treatment and/or (2) who were unsuitable for or unwilling to receive docetaxin [123]. From these, it is suggested that the treatment with Radium-223, which was based on a randomized controlled trial, might be useful for the patients with mCRPC, and that it is probable AT1/2R blockade might not be the best option for such patients. Intriguingly, although Hiroji Uemura et al. reported that blocking AT2R triggered the inhibition of proliferation of PCa cells [124], there was no randomized controlled trials reported as consequence. Furthermore, an ecologic study confirmed that there was no significant increase in the overall or site-specific cancer risk from ARBs, based on 15 trials enrolling 138 769 individuals [125]. Altogether, it is proposed that the alternative mechanisms of action might have played a key role in affecting the cancer risk; more importantly, it might perhaps explain the inconclusive findings on cancer risk in many trials of RAS blockade [43] .
|
Table 1: The summary of the effects of the Ang II/AT1R and AT2R receptor antagonists/blockers in PCa cells. |
|||
|
Ang II/AT1R and AT2R receptor antagonists/blockers |
Specific targets |
PCa cell lines |
Pharmacological effects |
|
Candesartan |
AT1R antagonist |
PC3, Du145, LNCaP |
① Inhibition of (1) cell growth and proliferation of PCa cells, following stimulation with either EGF or Ang II via the mechanism underlying the reduction of MAPK or STAT3 phosphorylation. [58] ② Alleviation of tumor angiogenesis in athymic nude mice [58] ③ Inhibition of growth of metastatic PC3 cell line [53] |
|
Fimasartan, Losartan, Eprosartan and Valsartan
|
AT2R blockers |
PC3, DU-145 and LNCap-LN3 |
① Reduction of PCa cell viability [59] ② Induction of autophagy-induced cell death in PC3 cell line. [59] ③ Fimasartan-induced inhibition of the migratory capacity of DU-145 and PC-3 cell lines [59] ④ Losartan-induced suppression of the growth and the proliferation of DU-145 cell line, via abolishing the Ang-II-induced PAX2 expression [60] |
|
Captopril |
ACE blocker |
Male patients aged 50-79 years old (clinical trials) |
① Showed a lower risk of developing and progressing PCa [61] |
|
Telmisartan |
|
PC3, DU-145 and LNCaP |
① Growth inhibition of PCa cells [62] ② Induction of early apoptisis in PCa cells. [62] ③ Suppression of PCa cell growth via inhibiting the Ang II-stimulated MAPK phosphorylation in LNCaP cell line [63]. |
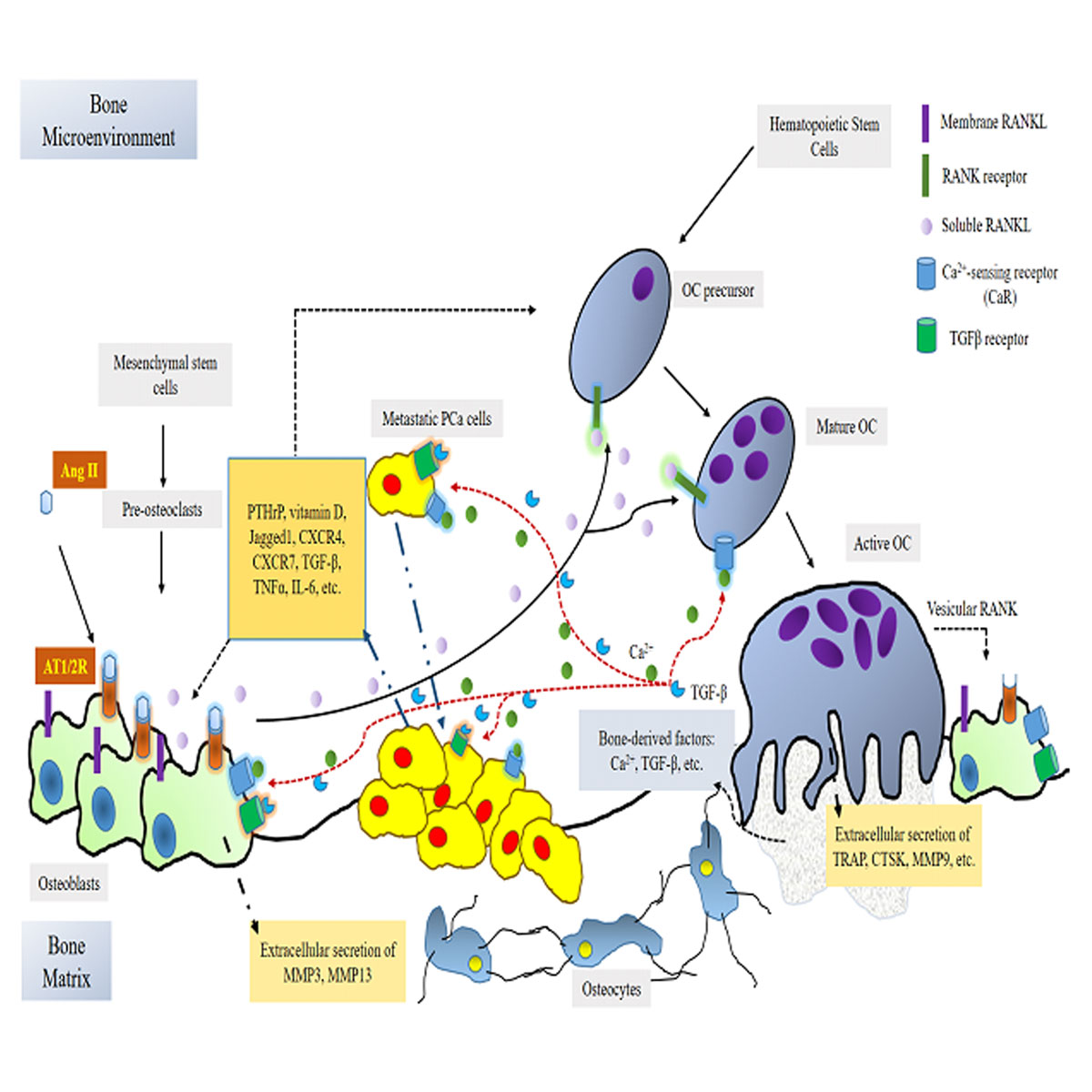 Figure 1. The possible mechanism underlying the RAS-mediated regulation of PCa bone metastasis. Ang II that is produced locally by the vascular endothelial binds to osteoblastic AT1R and AT2R, subsequently promoting OBs to the Cbfa1/Runx2-mediated secretion of the soluble RANKL into bone milieu. The OB-induced secretion of soluble RANKL binds and stimulates OC differentiation, thereby enhancing bone-resorbing activity of active OCs via the secretion of the specific enzymes (TRAP, CTSK and MMP9) into the bone matrix. Ca2+ ions and TFGβ, the bone elements, released from the OC-induced bone resorption, bind to CaRs and TFGβRs abundantly present in OBs, OCs and PCa cells, promoting the OB and OC differentiation and PCa bone metastasis. Several of the crucial factors, PTHrP, vitamin D, Jagged1, TFGβ, TNFα, etc., secreted by PCa cells, in turn, accelerate OB and OC differentiation. In addition, the active OC-secreted vesicular RANK further enhanced OB differentiation and then bone resorption.
Figure 1. The possible mechanism underlying the RAS-mediated regulation of PCa bone metastasis. Ang II that is produced locally by the vascular endothelial binds to osteoblastic AT1R and AT2R, subsequently promoting OBs to the Cbfa1/Runx2-mediated secretion of the soluble RANKL into bone milieu. The OB-induced secretion of soluble RANKL binds and stimulates OC differentiation, thereby enhancing bone-resorbing activity of active OCs via the secretion of the specific enzymes (TRAP, CTSK and MMP9) into the bone matrix. Ca2+ ions and TFGβ, the bone elements, released from the OC-induced bone resorption, bind to CaRs and TFGβRs abundantly present in OBs, OCs and PCa cells, promoting the OB and OC differentiation and PCa bone metastasis. Several of the crucial factors, PTHrP, vitamin D, Jagged1, TFGβ, TNFα, etc., secreted by PCa cells, in turn, accelerate OB and OC differentiation. In addition, the active OC-secreted vesicular RANK further enhanced OB differentiation and then bone resorption.
Accumulating clues has revealed the specific roles of the local RAS in regulating bone homeostasis and PCa bone metastasis; nonetheless, its specific roles are sometimes controversial. As a consequence, more works are necessarily carried out to further understand the pathophysiological role of the local RAS in bone metastases and the mechanisms underlying the possible therapeutic and/or adverse effects of angiotensin-converting enzyme inhibitors (ACEIs) and ARBs observed in preclinical and, particularly, in clinical trials. Also, it is essential to identify the downstream signaling pathways that stimulate AT1R and AT2R in both the progression of metastatic PCa and PCa bone mestastasis.
In summary, many published resports have confirmed a central role of the RAS in the development and the progession of PCa bone metastasis as elaborately outlined in this review article. Notably, ACEIs and ARBs are first-choice drugs used to treat the patients with the PCa bone metastasis, a large number of the crucial tasks that need to be clarified as follow: which is the most effective way to alleviate the local bone RAS? What is the role of the ACE2-Ang (1-7) pathway? For improving the current knowledge of the role of RAS in augmenting the PCa bone metastasis, more works would probably be prerequisite for further elucidating its functional roles in mediating the interplay of metastatic PCa cells and immune cells.
Ethical policy
Not applicable.
Author contributions
The author contributed solely to the work.
Competing interests
The author declares that there are no conflicts of interest.
Funding
Not applicable.
- Mirabito Colafella KM, Bovée DM, Danser AHJ: The renin-angiotensin-aldosterone system and its therapeutic targets. Exp Eye Res 2019, 186: 107680.
- Friis UG, Jensen BL, Sethi S, Andreasen D, Hansen PB, Skøtt O: Control of renin secretion from rat juxtaglomerular cells by cAMP-specific phosphodiesterases. Circ Res 2002, 90(9): 996-1003.
- Ma TK, Kam KK, Yan BP, Lam YY: Renin-angiotensin-aldosterone system blockade for cardiovascular diseases: current status. Br J Pharmacol 2010, 160(6): 1273-1292.
- Ryan JW, Ryan US, Schultz DR, Whitaker C, Chung A: Subcellular localization of pulmonary antiotensin-converting enzyme (kininase II). Biochem J 1975, 146(2): 497-499.
- Serfozo P, Wysocki J, Gulua G, Schulze A, Ye M, Liu P, Jin J, Bader M, Myöhänen T, García-Horsman JA et al: Ang II (Angiotensin II) Conversion to Angiotensin-(1-7) in the Circulation Is POP (Prolyloligopeptidase)-Dependent and ACE2 (Angiotensin-Converting Enzyme 2)-Independent. Hypertension 2020, 75(1): 173-182.
- Wright JW, Mizutani S, Harding JW: Focus on Brain Angiotensin III and Aminopeptidase A in the Control of Hypertension. International journal of hypertension 2012, 2012: 124758-124758.
- Saulière A, Bellot M, Paris H, Denis C, Finana F, Hansen JT, Altié MF, Seguelas MH, Pathak A, Hansen JL et al: Deciphering biased-agonism complexity reveals a new active AT1 receptor entity. Nat Chem Biol 2012, 8(7): 622-630.
- Ohtsu H, Suzuki H, Nakashima H, Dhobale S, Frank GD, Motley ED, Eguchi S: Angiotensin II signal transduction through small GTP-binding proteins: mechanism and significance in vascular smooth muscle cells. Hypertension 2006, 48(4): 534-540.
- Paing MM, Stutts AB, Kohout TA, Lefkowitz RJ, Trejo J: beta -Arrestins regulate protease-activated receptor-1 desensitization but not internalization or Down-regulation. J Biol Chem 2002, 277(2): 1292-1300.
- Cao Y, Kumar S, Namkung Y, Gagnon L, Cho A, Laporte SA: Angiotensin II type 1 receptor variants alter endosomal receptor-β-arrestin complex stability and MAPK activation. J Biol Chem 2020, 295(38):13169-13180.
- Becker BN, Kondo S, Chen JK, Harris RC: Tyrosine kinase inhibition affects type 1 angiotensin II receptor internalization. J Recept Signal Transduct Res 1999, 19(6): 975-993.
- He L, Du J, Chen Y, Liu C, Zhou M, Adhikari S, Rubin DT, Pekow J, Li YC: Renin-angiotensin system promotes colonic inflammation by inducing T(H)17 activation via JAK2/STAT pathway. Am J Physiol Gastrointest Liver Physiol 2019, 316(6): G774-g784.
- Chao Y, Ye P, Zhu L, Kong X, Qu X, Zhang J, Luo J, Yang H, Chen S: Low shear stress induces endothelial reactive oxygen species via the AT1R/eNOS/NO pathway. J Cell Physiol 2018, 233(2): 1384-1395.
- Hermosilla T, Encina M, Morales D, Moreno C, Conejeros C, Alfaro-Valdés HM, Lagos-Meza F, Simon F, Altier C, Varela D: Prolonged AT(1)R activation induces Ca(V)1.2 channel internalization in rat cardiomyocytes. Scientific reports 2017, 7(1): 10131-10131.
- Schleifenbaum J, Kassmann M, Szijártó IA, Hercule HC, Tano JY, Weinert S, Heidenreich M, Pathan AR, Anistan YM, Alenina N et al: Stretch-activation of angiotensin II type 1a receptors contributes to the myogenic response of mouse mesenteric and renal arteries. Circ Res 2014, 115(2): 263-272.
- Zhou Y, Castonguay P, Sidhom EH, Clark AR, Dvela-Levitt M, Kim S, Sieber J, Wieder N, Jung JY, Andreeva S et al: A small-molecule inhibitor of TRPC5 ion channels suppresses progressive kidney disease in animal models. Science 2017, 358(6368): 1332-1336.
- Yin G, Yan C, Berk BC: Angiotensin II signaling pathways mediated by tyrosine kinases. The International Journal of Biochemistry & Cell Biology 2003, 35(6): 780-783.
- O'Brien SL, Johnstone EKM, Devost D, Conroy J, Reichelt ME, Purdue BW, Ayoub MA, Kawai T, Inoue A, Eguchi S et al: BRET-based assay to monitor EGFR transactivation by the AT(1)R reveals G(q/11) protein-independent activation and AT(1)R-EGFR complexes. Biochem Pharmacol 2018, 158: 232-242.
- Roelle S, Grosse R, Aigner A, Krell HW, Czubayko F, Gudermann T: Matrix metalloproteinases 2 and 9 mediate epidermal growth factor receptor transactivation by gonadotropin-releasing hormone. J Biol Chem 2003, 278(47): 47307-47318.
- Rivas-Santisteban R, Lillo J, Muñoz A, Rodríguez-Pérez AI, Labandeira-García JL, Navarro G, Franco R: Novel Interactions Involving the Mas Receptor Show Potential of the Renin-Angiotensin system in the Regulation of Microglia Activation: Altered Expression in Parkinsonism and Dyskinesia. Neurotherapeutics 2021, https://doi.org/10.1007/s13311-020-00986-4. Epub ahead of print: 1-19.
- Gomes I, Sierra S, Devi LA: Detection of Receptor Heteromerization Using In Situ Proximity Ligation Assay. Curr Protoc Pharmacol 2016, 75: 2.16.11-12.16.31.
- Hadjidakis DJ, Androulakis, II: Bone remodeling. Ann N Y Acad Sci 2006, 1092: 385-396.
- Rodan GA: Bone homeostasis. Proceedings of the National Academy of Sciences of the United States of America 1998, 95(23): 13361-13362.
- Ortiz A, Lin SH: Osteolytic and osteoblastic bone metastases: two extremes of the same spectrum? Recent Results Cancer Res 2012, 192: 225-233.
- Rahim F, Hajizamani S, Mortaz E, Ahmadzadeh A, Shahjahani M, Shahrabi S, Saki N: Molecular Regulation of Bone Marrow Metastasis in Prostate and Breast Cancer. Bone Marrow Research 2014, 2014: 405920.
- Johnson RW, Suva LJ: Hallmarks of Bone Metastasis. Calcif Tissue Int 2018, 102(2): 141-151.
- Stangelberger A, Waldert M, Djavan B: Prostate cancer in elderly men. Reviews in urology 2008, 10(2): 111-119.
- Kawai AT, Martinez D, Saltus CW, Vassilev ZP, Soriano-Gabarró M, Kaye JA: Incidence of Skeletal-Related Events in Patients with Castration-Resistant Prostate Cancer: An Observational Retrospective Cohort Study in the US. Prostate Cancer 2019, 2019: 5971615.
- Garcia P, Schwenzer S, Slotta JE, Scheuer C, Tami AE, Holstein JH, Histing T, Burkhardt M, Pohlemann T, Menger MD: Inhibition of angiotensin-converting enzyme stimulates fracture healing and periosteal callus formation - role of a local renin-angiotensin system. Br J Pharmacol 2010, 159(8): 1672-1680.
- Gu SS, Zhang Y, Li XL, Wu SY, Diao TY, Hai R, Deng HW: Involvement of the skeletal renin-angiotensin system in age-related osteoporosis of ageing mice. Biosci Biotechnol Biochem 2012, 76(7): 1367-1371.
- Hagiwara H, Hiruma Y, Inoue A, Yamaguchi A, Hirose S: Deceleration by angiotensin II of the differentiation and bone formation of rat calvarial osteoblastic cells. J Endocrinol 1998, 156(3): 543-550.
- Lamparter S, Kling L, Schrader M, Ziegler R, Pfeilschifter J: Effects of angiotensin II on bone cells in vitro. J Cell Physiol 1998, 175(1): 89-98.
- Isaacs JT: Prostatic structure and function in relation to the etiology of prostatic cancer. The Prostate 1983, 4(4): 351-366.
- Yoshiji H, Yoshii J, Ikenaka Y, Noguchi R, Yanase K, Tsujinoue H, Imazu H, Fukui H: Suppression of the renin-angiotensin system attenuates vascular endothelial growth factor-mediated tumor development and angiogenesis in murine hepatocellular carcinoma cells. Int J Oncol 2002, 20(6): 1227-1231.
- Fujita M, Hayashi I, Yamashina S, Itoman M, Majima M: Blockade of angiotensin AT1a receptor signaling reduces tumor growth, angiogenesis, and metastasis. Biochemical and Biophysical Research Communications 2002, 294(2): 441-447.
- Masamune A, Hamada S, Kikuta K, Takikawa T, Miura S, Nakano E, Shimosegawa T: The angiotensin II type I receptor blocker olmesartan inhibits the growth of pancreatic cancer by targeting stellate cell activities in mice. Scand J Gastroenterol 2013, 48(5): 602-609.
- Rivera E, Arrieta O, Guevara P, Duarte-Rojo A, Sotelo J: AT1 receptor is present in glioma cells; its blockage reduces the growth of rat glioma. Br J Cancer 2001, 85(9): 1396-1399.
- DeAngelis LM: Brain tumors. N Engl J Med 2001, 344(2): 114-123.
- Suganuma T, Ino K, Shibata K, Kajiyama H, Nagasaka T, Mizutani S, Kikkawa F: Functional expression of the angiotensin II type 1 receptor in human ovarian carcinoma cells and its blockade therapy resulting in suppression of tumor invasion, angiogenesis, and peritoneal dissemination. Clin Cancer Res 2005, 11(7): 2686-2694.
- Huang W, Wu YL, Zhong J, Jiang FX, Tian XL, Yu LF: Angiotensin II type 1 receptor antagonist suppress angiogenesis and growth of gastric cancer xenografts. Dig Dis Sci 2008, 53(5): 1206-1210.
- Huang W, Yu LF, Zhong J, Qiao MM, Jiang FX, Du F, Tian XL, Wu YL: Angiotensin II type 1 receptor expression in human gastric cancer and induces MMP2 and MMP9 expression in MKN-28 cells. Dig Dis Sci 2008, 53(1): 163-168.
- Ni L, Feng Y, Wan H, Ma Q, Fan L, Qian Y, Li Q, Xiang Y, Gao B: Angiotensin-(1-7) inhibits the migration and invasion of A549 human lung adenocarcinoma cells through inactivation of the PI3K/Akt and MAPK signaling pathways. Oncol Rep 2012, 27(3): 783-790.
- Soto-Pantoja DR, Menon J, Gallagher PE, Tallant EA: Angiotensin-(1-7) inhibits tumor angiogenesis in human lung cancer xenografts with a reduction in vascular endothelial growth factor. Mol Cancer Ther 2009, 8(6): 1676-1683.
- Li H, Qi Y, Li C, Braseth LN, Gao Y, Shabashvili AE, Katovich MJ, Sumners C: Angiotensin type 2 receptor-mediated apoptosis of human prostate cancer cells. Mol Cancer Ther 2009, 8(12): 3255-3265.
- Chow L, Rezmann L, Imamura K, Wang L, Catt K, Tikellis C, Louis WJ, Frauman AG, Louis SN: Functional angiotensin II type 2 receptors inhibit growth factor signaling in LNCaP and PC3 prostate cancer cell lines. Prostate 2008, 68(6): 651-660.
- Fujiyama S, Matsubara H, Nozawa Y, Maruyama K, Mori Y, Tsutsumi Y, Masaki H, Uchiyama Y, Koyama Y, Nose A et al: Angiotensin AT(1) and AT(2) receptors differentially regulate angiopoietin-2 and vascular endothelial growth factor expression and angiogenesis by modulating heparin binding-epidermal growth factor (EGF)-mediated EGF receptor transactivation. Circ Res 2001, 88(1): 22-29.
- Li X, Lee JW, Graves LM, Earp HS: Angiotensin II stimulates ERK via two pathways in epithelial cells: protein kinase C suppresses a G-protein coupled receptor-EGF receptor transactivation pathway. Embo j 1998, 17(9): 2574-2583.
- Bedecs K, Elbaz N, Sutren M, Masson M, Susini C, Strosberg AD, Nahmias C: Angiotensin II type 2 receptors mediate inhibition of mitogen-activated protein kinase cascade and functional activation of SHP-1 tyrosine phosphatase. Biochem J 1997, 325 ( Pt 2)(Pt 2): 449-454.
- Ishizuka T, Goshima H, Ozawa A, Watanabe Y: Effect of angiotensin II on proliferation and differentiation of mouse induced pluripotent stem cells into mesodermal progenitor cells. Biochem Biophys Res Commun 2012, 420(1): 148-155.
- Kotha A, Sekharam M, Cilenti L, Siddiquee K, Khaled A, Zervos AS, Carter B, Turkson J, Jove R: Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol Cancer Ther 2006, 5(3): 621-629.
- Lee M-H, Kundu JK, Keum Y-S, Cho Y-Y, Surh Y-J, Choi BY: Resveratrol Inhibits IL-6-Induced Transcriptional Activity of AR and STAT3 in Human Prostate Cancer LNCaP-FGC Cells. Biomolecules & therapeutics 2014, 22(5): 426-430.
- Alhusban A, Al-Azayzih A, Goc A, Gao F, Fagan SC, Somanath PR: Clinically relevant doses of candesartan inhibit growth of prostate tumor xenografts in vivo through modulation of tumor angiogenesis. J Pharmacol Exp Ther 2014, 350(3): 635-645.
- Piastowska-Ciesielska AW, Kozłowski M, Wagner W, Domińska K, Och?dalski T: Effect of an angiotensin II type 1 receptor blocker on caveolin-1 expression in prostate cancer cells. Arch Med Sci 2013, 9(4): 739-744.
- Becker-Santos DD, Guo Y, Ghaffari M, Vickers ED, Lehman M, Altamirano-Dimas M, Oloumi A, Furukawa J, Sharma M, Wang Y et al: Integrin-linked kinase as a target for ERG-mediated invasive properties in prostate cancer models. Carcinogenesis 2012, 33(12): 2558-2567.
- Farah E, Li C, Cheng L, Kong Y, Lanman NA, Pascuzzi P, Lorenz GR, Zhang Y, Ahmad N, Li L et al: NOTCH signaling is activated in and contributes to resistance in enzalutamide-resistant prostate cancer cells. J Biol Chem 2019, 294(21): 8543-8554.
- Li K, Zhan X, Sun J, Wang T, Dong H, Jing F, Li D, Cao Y, Liu Y, Wang L et al: Oldhamianoside II inhibits prostate cancer progression via regulation of EMT and the Wnt/β-catenin signaling pathway. Oncol Lett 2018, 15(6): 9457-9463.
- Weber MJ, Gioeli D: Ras signaling in prostate cancer progression. J Cell Biochem 2004, 91(1):13-25.
- Bosetti M, Zanardi L, Hench L, Cannas M: Type I collagen production by osteoblast-like cells cultured in contact with different bioactive glasses. J Biomed Mater Res A 2003, 64(1): 189-195.
- Boonrungsiman S, Gentleman E, Carzaniga R, Evans ND, McComb DW, Porter AE, Stevens MM: The role of intracellular calcium phosphate in osteoblast-mediated bone apatite formation. Proc Natl Acad Sci U S A 2012, 109(35): 14170-14175.
- Rutkovskiy A, Stensløkken K-O, Vaage IJ: Osteoblast Differentiation at a Glance. Medical science monitor basic research 2016, 22:95-106.
- Soysa NS, Alles N: Osteoclast function and bone-resorbing activity: An overview. Biochem Biophys Res Commun 2016, 476(3): 115-120.
- Boyle WJ, Simonet WS, Lacey DL: Osteoclast differentiation and activation. Nature 2003, 423(6937): 337-342.
- Park JH, Lee NK, Lee SY: Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol Cells 2017, 40(10): 706-713.
- Cawley KM, Bustamante-Gomez NC, Guha AG, MacLeod RS, Xiong J, Gubrij I, Liu Y, Mulkey R, Palmieri M, Thostenson JD et al: Local Production of Osteoprotegerin by Osteoblasts Suppresses Bone Resorption. Cell Rep 2020, 32(10): 108052.
- Carbone LD, Vasan S, Prentice RL, Harshfield G, Haring B, Cauley JA, Johnson KC: The renin-angiotensin aldosterone system and osteoporosis: findings from the Women's Health Initiative. Osteoporos Int 2019, 30(10): 2039-2056.
- Asaba Y, Ito M, Fumoto T, Watanabe K, Fukuhara R, Takeshita S, Nimura Y, Ishida J, Fukamizu A, Ikeda K: Activation of renin-angiotensin system induces osteoporosis independently of hypertension. J Bone Miner Res 2009, 24(2): 241-250.
- Gu SS, Zhang Y, Wu SY, Diao TY, Gebru YA, Deng HW: Early molecular responses of bone to obstructive nephropathy induced by unilateral ureteral obstruction in mice. Nephrology (Carlton) 2012, 17(8): 767-773.
- Diao TY, Pan H, Gu SS, Chen X, Zhang FY, Wong MS, Zhang Y: Effects of angiotensin-converting enzyme inhibitor, captopril, on bone of mice with streptozotocin-induced type 1 diabetes. J Bone Miner Metab 2014, 32(3): 261-270.
- Hiruma Y, Inoue A, Hirose S, Hagiwara H: Angiotensin II stimulates the proliferation of osteoblast-rich populations of cells from rat calvariae. Biochem Biophys Res Commun 1997, 230(1): 176-178.
- Shimizu H, Nakagami H, Osako MK, Hanayama R, Kunugiza Y, Kizawa T, Tomita T, Yoshikawa H, Ogihara T, Morishita R: Angiotensin II accelerates osteoporosis by activating osteoclasts. Faseb j 2008, 22(7): 2465-2475.
- Nakai K, Kawato T, Morita T, Yamazaki Y, Tanaka H, Tonogi M, Oki H, Maeno M: Angiotensin II suppresses osteoblastic differentiation and mineralized nodule formation via AT1 receptor in ROS17/2.8 cells. Archives of medical science : AMS 2015, 11(3): 628-637.
- Hatton R, Stimpel M, Chambers TJ: Angiotensin II is generated from angiotensin I by bone cells and stimulates osteoclastic bone resorption in vitro. J Endocrinol 1997, 152(1): 5-10.
- Nakai K, Kawato T, Morita T, Yamazaki Y, Tanaka H, Tonogi M, Oki H, Maeno M: Angiotensin II suppresses osteoblastic differentiation and mineralized nodule formation via AT1 receptor in ROS17/2.8 cells. Arch Med Sci 2015, 11(3): 628-637.
- Li Y, Sikes RA, Malaeb BS, Yeung F, Law A, Graham SE, Pei M, Kao C, Nelson J, Koeneman KS et al: Osteoblasts can stimulate prostate cancer growth and transcriptionally down-regulate PSA expression in cell line models. Urol Oncol 2011, 29(6): 802-808.
- Gleave M, Hsieh JT, Gao CA, von Eschenbach AC, Chung LW: Acceleration of human prostate cancer growth in vivo by factors produced by prostate and bone fibroblasts. Cancer Res 1991, 51(14): 3753-3761.
- Sung SY, Hsieh CL, Law A, Zhau HE, Pathak S, Multani AS, Lim S, Coleman IM, Wu LC, Figg WD et al: Coevolution of prostate cancer and bone stroma in three-dimensional coculture: implications for cancer growth and metastasis. Cancer Res 2008, 68(23): 9996-10003.
- Lee YC, Cheng CJ, Bilen MA, Lu JF, Satcher RL, Yu-Lee LY, Gallick GE, Maity SN, Lin SH: BMP4 promotes prostate tumor growth in bone through osteogenesis. Cancer Res 2011, 71(15): 5194-5203.
- Özdemir BC, Hensel J, Secondini C, Wetterwald A, Schwaninger R, Fleischmann A, Raffelsberger W, Poch O, Delorenzi M, Temanni R et al: The molecular signature of the stroma response in prostate cancer-induced osteoblastic bone metastasis highlights expansion of hematopoietic and prostate epithelial stem cell niches. PLoS One 2014, 9(12): e114530.
- Schneider A, Kalikin LM, Mattos AC, Keller ET, Allen MJ, Pienta KJ, McCauley LK: Bone turnover mediates preferential localization of prostate cancer in the skeleton. Endocrinology 2005, 146(4): 1727-1736.
- Thudi NK, Martin CK, Murahari S, Shu ST, Lanigan LG, Werbeck JL, Keller ET, McCauley LK, Pinzone JJ, Rosol TJ: Dickkopf-1 (DKK-1) stimulated prostate cancer growth and metastasis and inhibited bone formation in osteoblastic bone metastases. Prostate 2011, 71(6): 615-625.
- Franceschi RT, Xiao G, Jiang D, Gopalakrishnan R, Yang S, Reith E: Multiple signaling pathways converge on the Cbfa1/Runx2 transcription factor to regulate osteoblast differentiation. Connect Tissue Res 2003, 44 Suppl 1(Suppl 1): 109-116.
- Kubalak SW, Webb JG: Angiotensin II enhancement of hormone-stimulated cAMP formation in cultured vascular smooth muscle cells. Am J Physiol 1993, 264(1 Pt 2): H86-96.
- Guan XX, Zhou Y, Li JY: Reciprocal roles of angiotensin II and Angiotensin II Receptors Blockade (ARB) in regulating Cbfa1/RANKL via cAMP signaling pathway: possible mechanism for hypertension-related osteoporosis and antagonistic effect of ARB on hypertension-related osteoporosis. Int J Mol Sci 2011, 12(7): 4206-4213.
- Dvorak-Ewell MM, Chen TH, Liang N, Garvey C, Liu B, Tu C, Chang W, Bikle DD, Shoback DM: Osteoblast extracellular Ca2+ -sensing receptor regulates bone development, mineralization, and turnover. J Bone Miner Res 2011, 26(12): 2935-2947.
- Yoshida N, Sato T, Kobayashi K, Okada Y: High extracellular Ca2+ and Ca2+-sensing receptor agonists activate nonselective cation conductance in freshly isolated rat osteoclasts. Bone 1998, 22(5): 495-501.
- Feng J, Xu X, Li B, Brown E, Farris AB, Sun S-Y, Yang JJ: Prostate cancer metastatic to bone has higher expression of the calcium-sensing receptor (CaSR) than primary prostate cancer. Receptors & clinical investigation 2014, 1(6): e270.
- Sanders JL, Chattopadhyay N, Kifor O, Yamaguchi T, Brown EM: Ca(2+)-sensing receptor expression and PTHrP secretion in PC-3 human prostate cancer cells. Am J Physiol Endocrinol Metab 2001, 281(6): E1267-1274.
- Mishra S, Tang Y, Wang L, deGraffenried L, Yeh IT, Werner S, Troyer D, Copland JA, Sun L-Z: Blockade of transforming growth factor-beta (TGFβ) signaling inhibits osteoblastic tumorigenesis by a novel human prostate cancer cell line. The Prostate 2011, 71(13): 1441-1454.
- Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, Massagué J, Mundy GR, Guise TA: TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest 1999, 103(2): 197-206.
- Santagata S, Ieranò C, Trotta AM, Capiluongo A, Auletta F, Guardascione G, Scala S: CXCR4 and CXCR7 Signaling Pathways: A Focus on the Cross-Talk Between Cancer Cells and Tumor Microenvironment. Front Oncol 2021, 11: 591386.
- Santagata S, Demichelis F, Riva A, Varambally S, Hofer MD, Kutok JL, Kim R, Tang J, Montie JE, Chinnaiyan AM et al: JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res 2004, 64(19): 6854-6857.
- Pajcini KV, Speck NA, Pear WS: Notch signaling in mammalian hematopoietic stem cells. Leukemia 2011, 25(10): 1525-1532.
- Tran MT: Overview of Ca2+ signaling in lung cancer progression and metastatic lung cancer with bone metastasis. Exploration of Targeted Anti-tumor Therapy 2021, https://doi.org/10.37349/etat.2021.00045. Epub ahead of print.
- Wu JY, Purton LE, Rodda SJ, Chen M, Weinstein LS, McMahon AP, Scadden DT, Kronenberg HM: Osteoblastic regulation of B lymphopoiesis is mediated by Gs{alpha}-dependent signaling pathways. Proc Natl Acad Sci U S A 2008, 105(44): 16976-16981.
- Ono T, Hayashi M, Sasaki F, Nakashima T: RANKL biology: bone metabolism, the immune system, and beyond. Inflamm Regen 2020, 40: 2.
- Penno H, Nilsson O, Brändström H, Winqvist O, Ljunggren O: Expression of RANK-ligand in prostate cancer cell lines. Scand J Clin Lab Invest 2009, 69(1): 151-155.
- Udagawa N, Takahashi N, Jimi E, Matsuzaki K, Tsurukai T, Itoh K, Nakagawa N, Yasuda H, Goto M, Tsuda E et al: Osteoblasts/stromal cells stimulate osteoclast activation through expression of osteoclast differentiation factor/RANKL but not macrophage colony-stimulating factor: receptor activator of NF-kappa B ligand. Bone 1999, 25(5): 517-523.
- Danks L, Komatsu N, Guerrini MM, Sawa S, Armaka M, Kollias G, Nakashima T, Takayanagi H: RANKL expressed on synovial fibroblasts is primarily responsible for bone erosions during joint inflammation. Ann Rheum Dis 2016, 75(6): 1187-1195.
- Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y et al: Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med 2006, 203(12): 2673-2682.
- Chen G, Sircar K, Aprikian A, Potti A, Goltzman D, Rabbani SA: Expression of RANKL/RANK/OPG in primary and metastatic human prostate cancer as markers of disease stage and functional regulation. Cancer 2006, 107(2): 289-298.
- Ono T, Hayashi M, Sasaki F, Nakashima T: RANKL biology: bone metabolism, the immune system, and beyond. Inflammation and Regeneration 2020, 40(1): 2.
- Ikebuchi Y, Aoki S, Honma M, Hayashi M, Sugamori Y, Khan M, Kariya Y, Kato G, Tabata Y, Penninger JM et al: Coupling of bone resorption and formation by RANKL reverse signalling. Nature 2018, 561(7722): 195-200.
- Kohara H, Kitaura H, Fujimura Y, Yoshimatsu M, Morita Y, Eguchi T, Masuyama R, Yoshida N: IFN-γ directly inhibits TNF-α-induced osteoclastogenesis in vitro and in vivo and induces apoptosis mediated by Fas/Fas ligand interactions. Immunol Lett 2011, 137(1-2): 53-61.
- Gao Y, Grassi F, Ryan MR, Terauchi M, Page K, Yang X, Weitzmann MN, Pacifici R: IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest 2007, 117(1): 122-132.
- Sercan AlpÖ, Durlanik S, Schulz D, McGrath M, Grün JR, Bardua M, Ikuta K, Sgouroudis E, Riedel R, Zehentmeier S et al: Memory CD8(+) T cells colocalize with IL-7(+) stromal cells in bone marrow and rest in terms of proliferation and transcription. European journal of immunology 2015, 45(4): 975-987.
- Seol MA, Kim JH, Oh K, Kim G, Seo MW, Shin YK, Sim JH, Shin HM, Seo BY, Lee DS et al: Interleukin-7 Contributes to the Invasiveness of Prostate Cancer Cells by Promoting Epithelial-Mesenchymal Transition. Sci Rep 2019, 9(1): 6917.
- Weitzmann MN, Roggia C, Toraldo G, Weitzmann L, Pacifici R: Increased production of IL-7 uncouples bone formation from bone resorption during estrogen deficiency. J Clin Invest 2002, 110(11): 1643-1650.
- Weitzmann MN, Cenci S, Rifas L, Brown C, Pacifici R: Interleukin-7 stimulates osteoclast formation by up-regulating the T-cell production of soluble osteoclastogenic cytokines. Blood 2000, 96(5): 1873-1878.
- Buchwald ZS, Kiesel JR, Yang C, DiPaolo R, Novack DV, Aurora R: Osteoclast-induced Foxp3+ CD8 T-cells limit bone loss in mice. Bone 2013, 56(1): 163-173.
- Senthilkumar R, Lee H-W: CD137L- and RANKL-mediated reverse signals inhibit osteoclastogenesis and T lymphocyte proliferation. Immunobiology 2009, 214(2): 153-161.
- Bertram EM, Lau P, Watts TH: Temporal segregation of 4-1BB versus CD28-mediated costimulation: 4-1BB ligand influences T cell numbers late in the primary response and regulates the size of the T cell memory response following influenza infection. J Immunol 2002, 168(8): 3777-3785.
- Piqueras L, Kubes P, Alvarez A, O'Connor E, Issekutz AC, Esplugues JV, Sanz MJ: Angiotensin II induces leukocyte-endothelial cell interactions in vivo via AT(1) and AT(2) receptor-mediated P-selectin upregulation. Circulation 2000, 102(17): 2118-2123.
- Nataraj C, Oliverio MI, Mannon RB, Mannon PJ, Audoly LP, Amuchastegui CS, Ruiz P, Smithies O, Coffman TM: Angiotensin II regulates cellular immune responses through a calcineurin-dependent pathway. J Clin Invest 1999, 104(12): 1693-1701.
- Mateo T, Naim Abu Nabah Y, Losada M, Estellés R, Company C, Bedrina B, Cerdá-Nicolás JM, Poole S, Jose PJ, Cortijo J et al: A critical role for TNFalpha in the selective attachment of mononuclear leukocytes to angiotensin-II-stimulated arterioles. Blood 2007, 110(6): 1895-1902.
- Sagawa K, Nagatani K, Komagata Y, Yamamoto K: Angiotensin receptor blockers suppress antigen-specific T cell responses and ameliorate collagen-induced arthritis in mice. Arthritis Rheum 2005, 52(6): 1920-1928.
- Weidanz JA, Jacobson LM, Muehrer RJ, Djamali A, Hullett DA, Sprague J, Chiriva-Internati M, Wittman V, Thekkumkara TJ, Becker BN: ATR blockade reduces IFN-gamma production in lymphocytes in vivo and in vitro. Kidney Int 2005, 67(6):2134-2142.
- Kim W, Ryan CJ: Androgen Receptor Directed Therapies in Castration-Resistant Metastatic Prostate Cancer. Current Treatment Options in Oncology 2012, 13(2): 189-200.
- Labrie F: Combined blockade of testicular and locally made androgens in prostate cancer: A highly significant medical progress based upon intracrinology. The Journal of Steroid Biochemistry and Molecular Biology 2015, 145: 144-156.
- Thiery-Vuillemin A, Hvid Poulsen M, Lagneau E, Ploussard G, Birtle A, Dourthe LM, Beal-Ardisson D, Pintus E, Trepiakas R, Antoni L et al: Impact of abiraterone acetate plus prednisone or enzalutamide on fatigue and cognition in patients with metastatic castration-resistant prostate cancer: initial results from the observational AQUARiUS study. ESMO Open 2018, 3(5): e000397.
- Chi K, Hotte SJ, Joshua AM, North S, Wyatt AW, Collins LL, Saad F: Treatment of mCRPC in the AR-axis-targeted therapy-resistant state. Ann Oncol 2015, 26(10): 2044-2056.
- Conteduca V, Wetterskog D, Sharabiani MTA, Grande E, Fernandez-Perez MP, Jayaram A, Salvi S, Castellano D, Romanel A, Lolli C et al: Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: a multi-institution correlative biomarker study. Ann Oncol 2017, 28(7): 1508-1516.
- Conteduca V, Jayaram A, Romero-Laorden N, Wetterskog D, Salvi S, Gurioli G, Scarpi E, Castro E, Marin-Aguilera M, Lolli C et al: Plasma Androgen Receptor and Docetaxel for Metastatic Castration-resistant Prostate Cancer. Eur Urol 2019, 75(3): 368-373.
- Hoskin P, Sartor O, O'Sullivan JM, Johannessen DC, Helle SI, Logue J, Bottomley D, Nilsson S, Vogelzang NJ, Fang F et al: Efficacy and safety of radium-223 dichloride in patients with castration-resistant prostate cancer and symptomatic bone metastases, with or without previous docetaxel use: a prespecified subgroup analysis from the randomised, double-blind, phase 3 ALSYMPCA trial. Lancet Oncol 2014, 15(12): 1397-1406.
- Uemura H, Hasumi H, Kawahara T, Sugiura S, Miyoshi Y, Nakaigawa N, Teranishi J, Noguchi K, Ishiguro H, Kubota Y: Pilot study of angiotensin II receptor blocker in advanced hormone-refractory prostate cancer. Int J Clin Oncol 2005, 10(6): 405-410.
- Effects of telmisartan, irbesartan, valsartan, candesartan, and losartan on cancers in 15 trials enrolling 138,769 individuals. J Hypertens 2011, 29(4): 623-635.
Annals of urologic oncology
p-ISSN: 2617-7765, e-ISSN: 2617-7773
 Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.