
 Submit Manuscript
Submit Manuscript
Case Report | Open Access
A Case of Malignant Pheochromocytoma of the Adrenal Gland in a Young Female
Veda Murthy Reddy Pogula1, Ershad Hussain Galeti1, Kashinath Vasanth Thakare1, Rahul Nekkanti1, Sandeep Reddy1, Ayesha Galeti1
1Narayana medical college, Nellore, Andhra Pradesh, India.
Correspondence: Ershad Hussain Galeti (Narayana medical college, Nellore, Andhra Pradesh, India; Email: dr.ershadhussain@gmail.com).
Annals of Urologic Oncology 2021, 4(2): 48-55. https://doi.org/10.32948/auo.2021.12.20
Received: 25 Aug 2021 | Accepted: 06 Dec 2021 | Published online: 30 Dec 2021 
Background Pheochromocytoma is a tumour of the adrenal medulla, derived from catecholamine producing chromaffin cells. Malignant pheochromocytomas constitute 10–25% of all cases. These are difficult to diagnose microscopically. Therefore, malignant pheochromocytomas are diagnosed by the presence of local invasion or metastatic disease.
Case presentation We present a case of malignant Pheochromocytoma in a 20-year-old woman from south India with classic symptoms whose urinary metanephrines levels were elevated. After controlling the blood pressure preoperatively and laparoscopic right-sided adrenalectomy was performed. The Postoperative period was uneventful. Histopathology proved to be malignant pheochromocytoma with a PASS score of 16/20 and immunohistochemical staining was positive. DOTATATE PET/CT showed no evidence of disease anywhere else in the body.
Conclusion Malignant pheochromocytomas are rare tumor, so they pose a significant diagnostic and therapeutic challenge. Surgery is the mainstay of treatment. DOTATATE PET/CT helps in the localization of metastatic disease.
Key words hypertension, pheochromocytoma, malignant pheochromocytoma, hypertension, paragangliomas, Ga-68 DOTATATE, PET/CT
Pheochromocytoma diagnosis is predominantly established by 24-hour urinary metanephrines and normetanephrines levels [5]. MRI is the preferred investigation for localization because contrast material used in CT scans can sometimes provoke paroxysms. 123-I MIBG (Meta iodo benzyl guanidine) scan identifies almost ninety percent of the primary Pheochromocytomas and is essential for the detection of extra-adrenal tumours and metastasis [3].
Laparoscopic resection is the preferred treatment for Pheochromocytoma now a days [3]. If a tumour is larger than 8-10 cm or any radiological signs of malignancy are seen, then an open approach is considered [2]. Preoperatively it is very important to control the blood pressure because after induction of anaesthesia or manipulation of the adrenal or extra-adrenal tumours during the surgery may cause fluctuations in blood pressure [6]. The agent of choice is a long-acting alpha-adrenergic blocker, phenoxybenzamine [6]. Beta blockade with propranolol is usually given a few days before surgery [6]. The beta-blockade should not be used before the alpha blockade, as it will cause unopposed vasoconstriction and can lead to potentially calamitous hypertension [6].
Post-operative ICU monitoring should be done for 24 hours as hypovolaemia and hypoglycaemia may occur [7]. Lifelong yearly biochemical tests need to be performed to identify any recurrent metastatic tumors [7]. In patients with Pheochromocytoma which is undiagnosed, death may occur from myocardial infarction, cerebrovascular accidents during or immediately even after a minor surgery [2].
Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) can be used to assess for malignant potential. Features to assess are periadrenal adipose invasion, > 3 mitoses / high power fields, atypical mitoses, necrosis, cellular spindling, marked nuclear pleomorphism, cellular monotony, large nests or diffuse growth, high cellularity, capsular invasion, vascular invasion. Score ≥ 6 is concerning for malignancy (Table 1) [8].
Malignant pheochromocytomas constitute 10% of all cases [9]. Given the rarity of this condition, therapeutic options are not well defined and those which are available are mostly palliative [10]. Malignancy in pheochromocytoma is difficult to diagnose microscopically. Often malignant pheochromocytomas are diagnosed by the presence of local invasion or metastatic disease. Even though 123/131I-metaiodobenzylguanidine (123/131I-MIBG) scintigraphy is an excellent functional imaging tool in adrenal pheochromocytoma, it has demonstrated low sensitivity in extra-adrenal and metastatic pheochromocytoma [11]. The high sensitivity and accuracy in 68Ga-DOTATATE PET/CT imply that somatostatin receptor PET/CT imaging may offer a better overview on tumour spread and extension than 131I-MIBG and 18F-FDG. 68Ga-DOTATATE PET/CT should be recommended into the standard algorithm of evaluation such tumours [12]. When metastatic, the most frequent locations are bone, lymph nodes, liver, lung, and brain. Currently, no diagnostic molecular or histological marker exists for malignancy and most patients are diagnosed with malignant PPGL after they have developed unresectable disease and metastatic disease [13]. The 5-year survival without metastasis has been reported to be approximately 89.3% [14]. When metastasis is detected, the prognosis is usually poor, with a 5-year survival rate of only 60% [15]. Most of these cases then resort to systemic chemotherapy or radiopharmaceutical agents, such as metaiodobenzylguanidine. These therapies lead mostly to partial radiographic response, disease stabilization, and symptomatic control in about 40% of cases [13, 15].
In this article, we report a case of symptomatic right-sided malignant adrenal pheochromocytoma in a young woman who presented for investigation of infertility.
A 20-year-old woman who presented to our hospital with either no medical or family history of endocrine disease. She visited the department of Obstetrics and Gynaecology at our hospital for infertility treatment. The adrenal tumour was diagnosed using ultrasound and CECT abdomen, and subsequently, the patient was referred to the urologist for further treatment. The patient had the following symptoms: paroxysmal headache, palpitations and sweating associated with dizziness. On examination her pulse was 110 beats per minute, regular, good volume and blood pressure was 180/110 mm of Hg.
Abdominal CECT revealed a hypodense tumour (30 HU) of size 5 × 5 cm in the right suprarenal region with central necrotic areas and rapid washout, the edges were well defined and the tumour did not involve the adjoining tissues (Figure 1). Right pheochromocytoma was suspected, therefore 24 hourly urinary metanephrine, normetanephrine were assessed and the values were as follows: Normetanephrines-1285?g/L (reference ranges < 600 μg/24 h) and metanephrines-126 ?g/L (reference ranges < 26-312 μg/24 h). Thyroid-stimulating hormone and ultrasound imaging of thyroid and parathyroid glands were normal. Preoperatively blood pressure was controlled with alpha-blockers and beta-blockers were added one week before the surgery. The patient had consented to laparoscopic right-sided adrenalectomy. On laparoscopy, a mass of about 5×5 cm was found at the right suprarenal area displacing the right kidney. Para-aortic lymph nodes were not enlarged and the liver appeared normal. Total right laparoscopic adrenalectomy had been performed. The resected specimen weighed 200 gm (Figure 2). The patient's condition during surgery and postoperatively was stable. Post-operatively blood pressure was normal from day 1. Pathological examination revealed a large number of nuclear fissions in the tumour cells, with the PASS score of 16/20 (Table 2), which led to the diagnosis of malignant pheochromocytoma with invasion beyond the capsule (Figure 3). Immunohistochemical staining was positive for chromogranin A and synaptophysin. S-100 immunostain showed moderate nuclear and cytoplasmic positivity including sustentacular cells.
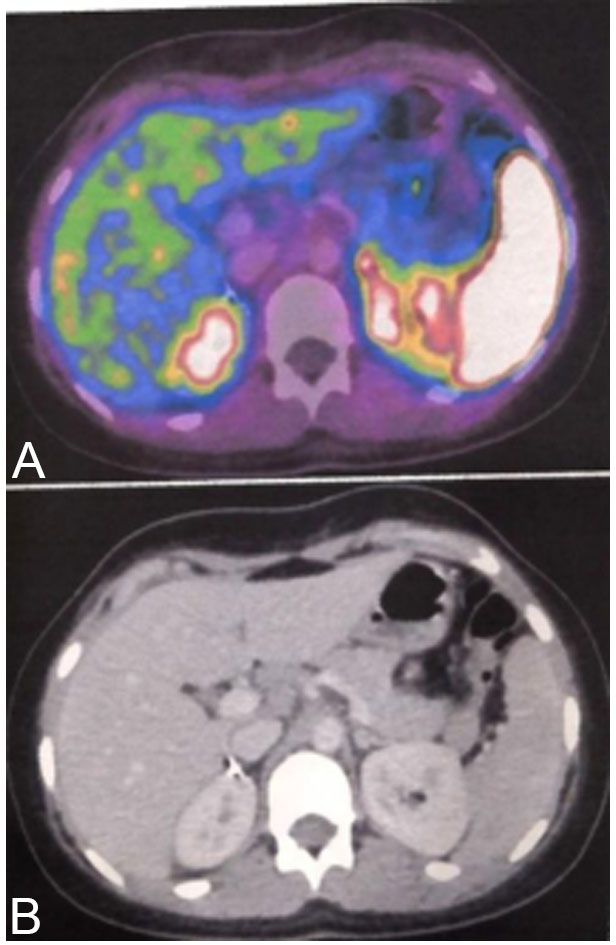
The patient recovered without any postoperative problems, and plasma catecholamine levels normalized. Examination by imaging revealed no residual tumour. 68Ga DOTATATE PET/CT showed no evidence of significant uptake suggestive of no disease anywhere else in the body (Figure 4). The patient is continuing fertility treatment and patient is currently adhering to strict follow-up as an outpatient at present. 1 year after the surgery, there are no signs of recurrence.
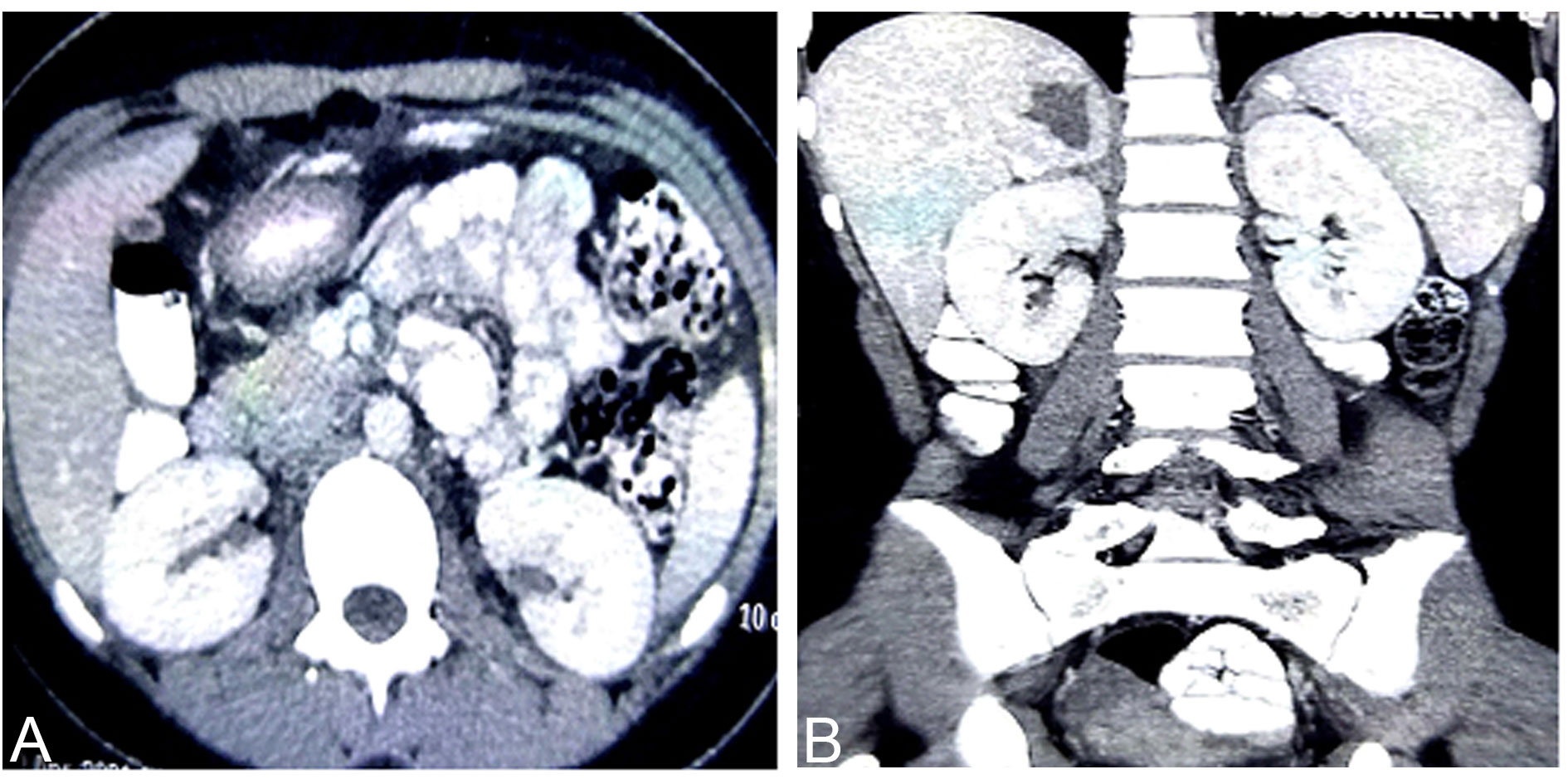 Figure 1. CT scan of the abdomen showing a mass in the right adrenal gland. (a) Axial view; (b) Coronal view.
Figure 1. CT scan of the abdomen showing a mass in the right adrenal gland. (a) Axial view; (b) Coronal view.
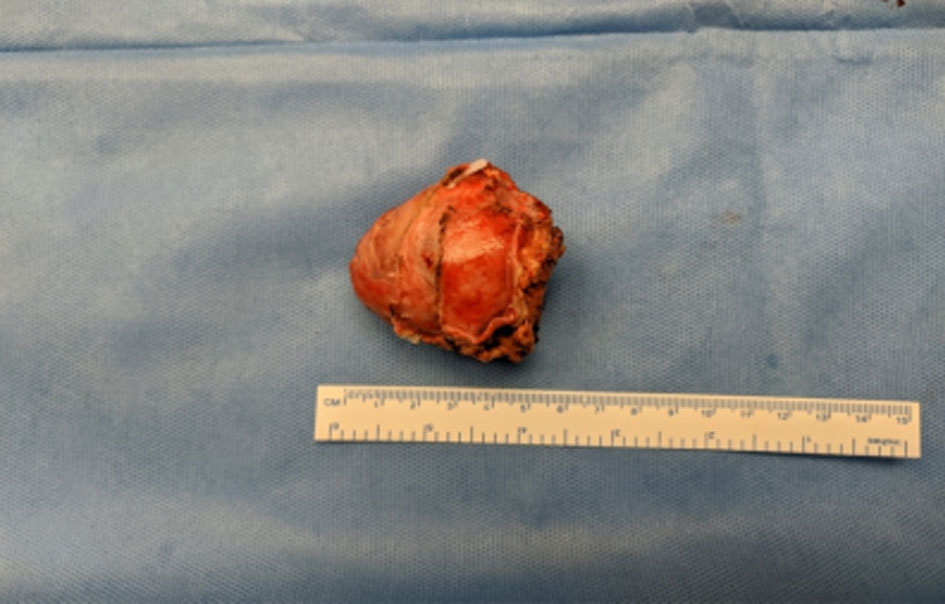 Figure 2. Macroscopic picture of the surgical specimen.
Figure 2. Macroscopic picture of the surgical specimen.
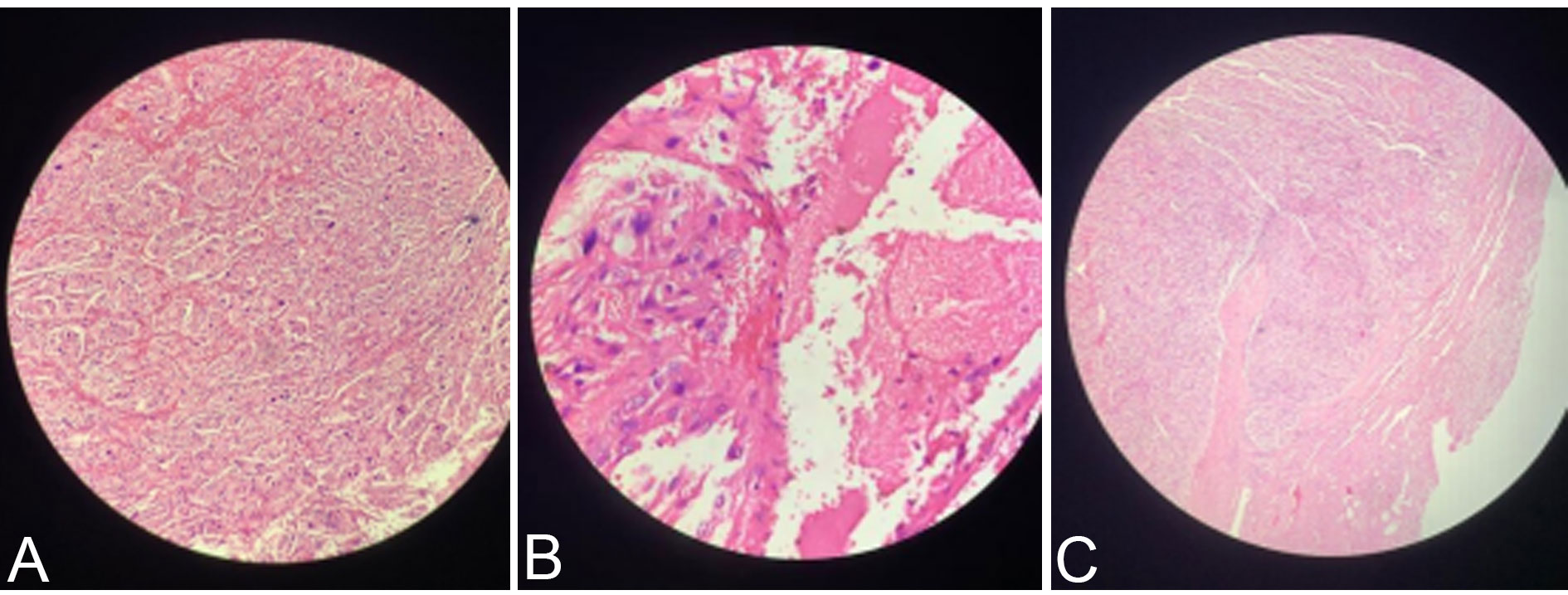 Figure 3. (a). Microscopically zellballen pattern of arrangement of tumour cells with mitotic activity; (b). Area of necrosis; (c). Capsular invasion of tumour.
Figure 3. (a). Microscopically zellballen pattern of arrangement of tumour cells with mitotic activity; (b). Area of necrosis; (c). Capsular invasion of tumour.
|
Table 1. Pheochromocytoma of the adrenal gland scoring. |
|
|
HISTOMORPHOLOGICAL PARAMETER |
Score |
|
Nuclear hyperchromasia |
1 |
|
Profound nuclear pleomorphism |
1 |
|
Capsular invasion |
1 |
|
Vascular invasion |
1 |
|
Extension into periadrenal adipose tissue |
2 |
|
Atypical mitotic figures |
2 |
|
Greater than 3 mitotic figures/10 high-power field |
2 |
|
Tumour cell spindling |
2 |
|
Cellular monotony |
2 |
|
High cellularity |
2 |
|
Central or confluent tumour necrosis |
2 |
|
Large nests or diffuse growth (>10% of tumour volume) |
2 |
|
Total |
20 |
|
Table 2. PASS score in our patient. |
|
|
HISTOMORPHOLOGICAL PARAMETER |
Score |
|
Nuclear hyperchromasia |
1 |
|
Profound nuclear pleomorphism |
1 |
|
Capsular invasion |
1 |
|
Vascular invasion |
1 |
|
Extension into periadrenal adipose tissue |
Absent |
|
Atypical mitotic figures |
Absent |
|
Greater than 3 mitotic figures/10 high-power field |
2 |
|
Tumour cell spindling |
2 |
|
Cellular monotony |
2 |
|
High cellularity |
2 |
|
Central or confluent tumour necrosis |
2 |
|
Large nests or diffuse growth (>10% of tumour volume) |
2 |
|
Total |
16 |
Pheochromocytoma is a tumour of the adrenal medulla and chromaffin tissues in the other parts of the body (paragangliomas). They secrete epinephrine or norepinephrine, resulting in sustained or episodic hypertension and other symptoms of catecholamine excess [16].
Approximately 0.2%-0.5% of patients are diagnosed with pheochromocytoma on hypertensive workup and accounts for about 5% of adrenal tumours incidentally discovered by CT scanning, whereas 10% of pheochromocytomas are found incidentally [6]. Most Pheochromocytomas occur sporadically, but they may be associated with various familial syndromes such as multiple endocrine neoplasia (MEN) 2A (medullary thyroid carcinoma, pheochromocytoma, and hyperparathyroidism), MEN 2B (medullary thyroid carcinoma, pheochromocytoma, mucosal neuromas, marfanoid habitus, and ganglioneuromatosis), von Recklinghausen disease, von Hippel- Lindau disease [7].
Essentials of diagnosis for Pheochromocytoma are hypertension, frequently sustained, with or without paroxysms, episodic headache, excessive sweating, palpitation, and visual blurring, postural tachycardia, elevated urinary catecholamines or their metabolites, hypermetabolism and hyperglycemia [16]. Our patient had sustained hypertension, headache, sweating and dizziness and her urinary normetanephrine levels were elevated.
Urinary output of metanephrines and/or free catecholamines is elevated in more than ninety-five percent of patients with pheochromocytoma. However many drugs and diets can interfere with the results of VMA. Zianni et al [17]. reported a case of pheochromocytoma with hypertension having normal urinary normetanephrine levels but the tumour was benign and in our case the tumour was malignant.
Approximately ten percent of pheochromocytomas are malignant. This rate is high in extra-adrenal tumours (paragangliomas). The diagnosis of malignancy implies metastasis of chromaffin tissue, most commonly to the bone, lymph nodes and liver [18, 19]. Histologically, pheochromocytomas are usually well-circumscribed masses. Nests of polygonal or rounded cells, eponymously named Zellballen, are characteristic. Features suggestive of aggressive tumour growth include spindle morphology, increased mitotic rate, and invasion of the organ's capsule. Cytological findings rarely are to determine whether a Pheochromocytoma is malignant or benign. The veins and capsules may also be invaded even in clinically benign tumors [20].
Malignancy can only be diagnosed in the presence of metastases or invasion into surrounding tissues [16]. In our case, the tumour was not invading the kidney. Several scoring systems considering invasion, histologic growth patterns, cytologic features, or mitotic activity have been proposed to calculate the risk of malignancy [21, 22]. One of the most utilized score is “Pheochromocytoma of the Adrenal gland Scales Score (PASS),” proposed by Thompson in 2002. A PASS score ≥4 was at first considered suggestive for a biological aggressive behaviour, but a later study revealed that all malignant PCCs had a PASS >6 [23]. On the basis of these results a PASS score <4 or="">6 suggest benign and malignant lesions respectively, whereas a value between 4 and 6 suggests an intermediate risk. The PASS score is based on the degree of tumor invasion, histologic growth patterns, cytologic features, or mitotic activity is used to distinguish a benign from a malignant PCC. Our patient’s PASS score was 16 with >3 mitoses/high power fields, necrosis, cellular spindling, marked nuclear pleomorphism, cellular monotony, large nests or diffuse growth, high cellularity, capsular invasion, vascular invasion out of a maximum score of 20, making it a malignant Pheochromocytoma (Table 2).
Further information can derive from the evaluation of specific molecular markers. Several malignancy tissue markers such as cyclooxygenase-2, secretogranin II-derived peptide, N-cadherin, vascular endothelial growth factor (VEGF), endothelin receptor type A (ETA), and type B (ETB) and telomerase have been identified. In particular telomerase, which is a ribonucleoprotein complex that includes the telomerase RNA component, the telomerase associated protein (TP1), the telomerase catalytic subunit (hTERT), and the heat shock protein 90 (HSP90) seem to be closely related to the malignant potential of paragangliomas (PGLs). In fact an upregulation of hTERT, HSP90, and telomerase activity has been evidenced in malignant cells of pheochromocytomas (PCCs) [24]. The Ki-67 nuclear antigen represents another potential molecular marker which has been associated with more aggressive cancers. A Ki-67 index >3% is considered a useful parameter predicting malignant potential [25]. Another promising marker predicting metastatic potential seems the transcription factor SNAIL. Positive immunostaining has been found significantly higher in metastatic than benign PGLs [26]. Novel biomarkers are recently being identified by micro-RNA expression profiling studies. Micro-RNA is small single-strand, nonprotein coding RNA fragments, which are able to negatively regulate protein expression by either cleavage or translational repression of mRNA [27]. Recently,Meyer-Rochow, and colleagues [28] investigated 12 malignant, 12 benign tumors, and 5 healthy adrenal medulla samples. They found that miR-483-5p was overexpressed, while miR15a and miR-16, which are involved in proliferation and apoptosis, were downregulated in malignant compared to benign tumors. MicroRNA expression is tissue specific, and it has been demonstrated to be altered in several other human tumors, For these reasons, they can be of great relevance for the establishment of malignancy, but further investigations in larger cohorts of patients are necessary to confirm these encouraging results.
Until 2000, only 10% of PGLs were considered of genetic origin and linked to hereditary syndromes: von Hippel Lindau disease (VHL), multiple endocrine neoplasia type 2 (MEN2) and neurofibromatosis type 1 (NF1), due respectively to a germ line mutation in tumor-suppressor gene VHL [29]. protooncogene RET [30] and tumor-suppressor gene NF1 [31]. In the last years it has been demonstrated that about 30% of the apparently sporadic PGLs are due to a germ-line mutation in one of the susceptibility genes [32]. This group of genes includes those encoding the four subunits (A, B, C, and D) of the succinate dehydrogenase (SDH) [33], the recently identified gene SDHAF2, which is responsible for the flavination of the SDHA subunit [34], and the very recently discovered TMEM127 [35] and MAX [36], both mainly related to bilateral PCCs. Germ line mutations in SDHA, SDHB, SDHC, SDHD, and SDHAF2 genes are responsible for the occurrence of syndromes named PGL5, PGL4, PGL3, PGL1, and PGL2, respectively; to note, SDHB-mutations are generally associated with higher morbidity and mortality than mutations in the other SDHx genes [37]. Overall, to date, 10 susceptibility PGLs genes have been identified, so that the initial 10% of cases classified as genetically determined has increased to 30%. Nevertheless, the number of the susceptibility genes is likely to increase. In fact, many young PGL patients, where the mutation frequency is higher, are still classified as sporadic, and some PGLs patients with a positive family history do not show any mutation in the so far known susceptibility genes.
About eight percent of patients with a benign Pheochromocytoma subsequently develop metastasis. Surgical excision is the only chance for cure. Even in patients with metastatic disease, tumour debulking can be considered to reduce the tumour burden and to control the catecholamine excess. Symptomatic treatment can be obtained with alpha-blockers. Mitotane can be used as adjuvant or palliative treatment [2]. Treatment with 131-I MIBG or combination chemotherapy has resulted in a partial response in thirty percent and an improvement of symptoms in eighty percent of patients. [68Ga]-DOTATATE PET/CT showed a significantly superior detection rate to all other functional and anatomical imaging modalities and may represent the preferred future imaging modality in the evaluation of malignant/ metastatic pheochromocytomas [39]. In our patient, DOTATATE PET/CT showed no evidence of significant uptake suggestive of no disease anywhere else in the body.
Understanding specific molecular pathways alteration responsible for malignant PGLs development might hopefully in the future lead to multiple molecular-targeted therapy for a successful treatment. Effectiveness of these therapies is due to a cytostatic effect, as they interfere with specific molecular targets found along the oncogenic signaling pathways responsible for carcinogenesis and tumor growth. As stated above, both benign and malignant PGLs gene mutations are part of two distinct molecular pathways leading to tumorigenesis: cluster 1 includes mutations of VHL, SDHB, and SDHD and is associated to pseudohypoxia and aberrant VEGF signaling, leading to abnormal hypoxia inducible factor (HIF) activation and overexpression of angiogenic factors, while cluster 2 includes mutations of RET, NF1, TMEM127, and MAX and is associated with abnormal activation of kinasesignaling pathways such as PI3kinase/AKT, RAS/RAF/ERK, and mTOR1/p70s6K, leading to abnormal cell growth and lack of apoptosis capacity. In addition, malignant PCCs seem to overexpress HSP90, a molecular chaperone that assists in folding proteins and stabilizes various oncoproteins that play a role in malignant phenotype [40, 41]. Thus, HIF1a inihbitors are molecular targeted drugs interfering with HIF hypoxia-driven transcription pathway, decreasing HIF activity directly, PX-478, and indirectly, PX-12 (1-methylpropyl 2-imidazolyl disulfide). These agents have shown marked antitumoral activity in human tumor xenografts in mice and seem to be promising also for malignant PGLs, but conclusive data are missing [42]. The mTOR inhibitor everolimus (RAD001) in combination with octreotide has been evaluated for low and intermediate grade neuroendocrine tumors, with good results [43]. The efficacy of everolimus has been evaluated also in malignant PGLs, but all patients experienced disease progression [44]. Maybe the low efficacy is due to a compensatory PI3K/AKT and ERK activation in response to mTOR inhibition, so a specific novel dual PI3k/mTOR inhibitor (NVP-BEZ235) might offer a novel therapeutic approach [45]. Further studies on the PI3K/AKT/mTOR pathway have to be conducted to find a more specific molecular target in its signalling. Several studies have demonstrated overexpression in malignant PCCs of angiogenic molecules, such as VEGF, its receptor, angiopoietin-2, and the endothelin receptors ETA and ETB46, leading a strong evidence that targeting this pathway with antiangiogenic therapies could represent a new promising treatment option. Accordingly, sunitinib, a receptor tyrosine kinase inhibitor acting on several targets (VEGF, PDGF, and c-KIT), with strong antiangiogenic and antitumor activity, has been used in the treatment of malignant PCCs, with promising results [47]. Imatinib, another tyrosine kinase inhibitor already used for hematologic and gastrointestinal stromal tumors, has not been found effective for malignant PCCs treatment [48]. Thalidomide, by targeting VEGF and basic fibroblast growth factor, is an antiangiogenic agent evaluated for treatment of metastatic renal cell cancer, multiple myeloma and nonsmall cell lung cancer [49]. It has been used in combination with Temozolomide in neuroendocrine tumors [50] obtaining an objective biochemical response rate in about 40% and a radiologic response rate in 33% of malignant PCCs, but lymphopenia occurred in about 70% of treated patients. Activators of prolyl hydroxylase (PHD) (such as ERBB2 inhibitors) are now on evaluation as promising antineoplastic therapies. These molecules decrease the expression levels of some angiogenic factors, such as VEGF, acting on HIF pathway, by activating the PHD, thus increasing HIF hydroxylation, and promoting its degradation [51]. The radiopharmaceutical metaiodobenzylguanidine (MIBG) was created in 1979 [52]. 131I-MIBG was used to treat malignant, non resectable or metastatic PCC. MIBG is labeled with 131I at the meta-position and is taken up by the noradrenaline transporter. Once inside the tumor cell, MIBG releases lethal radiation that causes severe DNA damage, inhibiting cell proliferation and causing cell death. Up to 80% of MPPG patients have tumors that express the noradrenaline transporter in the cell membrane [53]. Responses to MIBG are, however, limited, with only 30% of MPPG patients seeing a clinical benefit [54]. Adjuvant external beam radiotherapy was recommended after surgical debulking of PCC, especially in patients with incomplete or R1 resection [55]. Yu et al. [56] was able to reverse cranial nerve deficit due to PCC metastatic tumor in the parasellar region by giving 25 Gy, the patient suffered a local recurrence again 2 years later, and was again treated with 20 Gy, achieving prompt tumor response, the patient’s hepatic metastasis was controlled for >2 year by giving 32.4 Gy. They recommended a dose of 40-50 Gy to achieve a more long-lasting tumor control for inoperable tumor. Surveillance should include plasma-free or urinary metanephrines every 3 months for 12 months. Metanephrines should be checked annually thereafter. Lifelong surveillance is recommended. Most recurrences occur within 5 years, but recurrences have been reported as long as 40 years from original resection. Nonsurgical treatment includes high-dose MIBG or chemotherapy comprising vincristine, dacarbazine, and cyclophosphamide. Unfortunately, there have been no complete responses reported and only 57% of cases experience a partial response. The five-year survival rate is less than fifty percent in malignant pheochromocytoma [57].
Acknowledgements
We extend their sincere thanks to all the patients who participated in the study.
Ethical policy
Approval from institutional ethical committee was taken, the principles of the Helsinki Declaration were followed.
Author contributions
VMR- Supervision, Critical Review, Conception; EHG- Design, Materials, Data collection, Writing; KVT, RN, SR- Analysis and Interpretation; EHG, AG- Literature Review.
Competing interests
The authors declare no competing interests.
Funding
This study was financially not supported by any company.
- Eisenhofer G, Lenders JW, Goldstein DS, Mannelli M, Csako G, Walther MM, Brouwers FM, & Pacak K: Pheochromocytoma Catecholamine Phenotypes and Prediction of Tumor Size and Location by Use of Plasma Free Metanephrines. Clin Chem 2005, 51(4): 735-744.
- Ali KAS: Malignant Pheochromocytoma: A Case Report. Emerg Med: Open Access 2014: 04(05).
- Collins D. Bailey & Love’s Short Practice of Surgery. 25th Edn N. S. Williams, C. J. K. Bulstrode and P. R. O’Connell (eds) 283 × 225 mm. Pp. 1514. Illustrated. Hodder Arnold: London. Br J Surg 2008, 95(10): 1311-1311.
- Karagiannis A, Mikhailidis DP, Athyros VG, & Harsoulis F: Pheochromocytoma: an update on genetics and management. Endocr Relat Cancer 2007, 14(4), 935-956.
- Pacak K: Phaeochromocytoma: a catecholamine and oxidative stress disorder. Endocr Regul 2011, 45(2): 65-90.
- Zuber SM, Kantorovich V, & Pacak K: Hypertension in Pheochromocytoma: Characteristics and Treatment. Endocrinol Metab Clin North Am 2011, 40(2): 295-311.
- Därr R, Lenders JWM, Hofbauer LC, Naumann B, Bornstein SR, & Eisenhofer G: Pheochromocytoma - update on disease management. Ther Adv Endocrinol Metab 2012, 3(1): 11-26.
- Thompson LDR: Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) to Separate Benign From Malignant Neoplasms. Am J Surg Pathol 2002, 26(5): 551-566.
- Patel D, Phay JE, Yen TWF, Dickson PV, Wang TS, Garcia R, Yang AD, Solórzano CC, & Kim LT: Update on Pheochromocytoma and Paraganglioma from the SSO Endocrine/Head and Neck Disease-Site Work Group. Part 1 of 2: Advances in Pathogenesis and Diagnosis of Pheochromocytoma and Paraganglioma. Ann Surg Oncol 2020, 27(5): 1329-1337.
- Jimenez C, Rohren E, Habra M. A, Rich T, Jimenez P, Ayala-Ramirez M, & Baudin E: Current and Future Treatments for Malignant Pheochromocytoma and Sympathetic Paraganglioma. Curr Oncol Rep 2013, 15(4): 356-371.
- Wiseman GA, Pacak K, O’Dorisio MS, Neumann DR, Waxman AD, Mankoff DA, Heiba SI, Serafini AN, Tumeh SS, Khutoryansky et al: Usefulness of 123I-MIBG Scintigraphy in the Evaluation of Patients with Known or Suspected Primary or Metastatic Pheochromocytoma or Paraganglioma: Results from a Prospective Multicenter Trial. J Nucl Med 2009, 50(9): 1448-1454.
- Tan TH, Hussein Z, Saad FF, & Shuaib IL: Diagnostic Performance of (68)Ga-DOTATATE PET/CT, (18)F-FDG PET/CT and (131)I-MIBG Scintigraphy in Mapping Metastatic Pheochromocytoma and Paraganglioma. Nucl Med Mol Imaging 2005, 49(2): 143-151.
- Jimenez C, Rohren E, Habra M. A, Rich T, Jimenez P, Ayala-Ramirez M, & Baudin E: Current and Future Treatments for Malignant Pheochromocytoma and Sympathetic Paraganglioma. Curr Oncol Rep 2013, 15(4): 356-371.
- Korevaar T. I. M, & Grossman A. B: Pheochromocytomas and paragangliomas: assessment of malignant potential. Endocrine 2011, 40(3): 354-365.
- Jimenez P, Tatsui C, Jessop A, Thosani S, & Jimenez C: Treatment for Malignant Pheochromocytomas and Paragangliomas: 5 Years of Progress. Curr Oncol Rep 2017, 19(12): 83.
- Pasternak J. D, Seib C. D, Seiser N, Tyrell J. B, Liu C, Cisco R. M, Gosnell J. E, Shen W. T, Suh I, & Duh Q.Y. Differences Between Bilateral Adrenal Incidentalomas and Unilateral Lesions. JAMA Surg 2015, 150(10): 974.
- Zianni D, Tzanela M, Klimopoulos S, & Thalassinos N. C: Symptomatic pheochromocytoma with normal urinary catecholamine metabolites. Hormones (Athens) 2004, 3(2): 132-137.
- Zelinka T, Musil Z, Dušková J, Burton D, Merino M. J, Milosevic D, Widimský J, & Pacak K: Metastatic pheochromocytoma: Does the size and age matter? Eur J Clin Invest 2011, 41(10): 1121-1128.
- Garg MK, Kharb S, Brar KS: Gundgurthi A, & Mittal R: Medical management of pheochromocytoma: Role of the endocrinologist. Indian J Endocrinol Metab 2011, 15(Suppl4): S329-S336.
- Johnson MH, Cavallo JA, & Figenshau RS: Malignant and Metastatic Pheochromocytoma: Case Report and Review of the Literature. Urol Case Rep 2014, 2(4): 139-141.
- Ilona Linnoila R, Keiser HR, Steinberg SM, & Lack EE: Histopathology of benign versus malignant sympathoadrenal paragangliomas: Clinicopathologic study of 120 cases including unusual histologic features. Hum Pathol 1990, 21(11): 1168-1180.
- Kimura N, Watanabe T, Noshiro T, Shizawa S, & Miura Y: Histological Grading of Adrenal and Extra-adrenal Pheochromocytomas and Relationship to Prognosis: A Clinicopathological Analysis of 116 Adrenal Pheochromocytomas and 30 Extra-adrenal Sympathetic Paragangliomas Including 38 Malignant Tumors. Endocr Pathol 2005, 16(1): 023-032.
- Strong VE, Kennedy T, Al-Ahmadie H, Tang L, Coleman J, Fong Y, Brennan M, & Ghossein RA: Prognostic indicators of malignancy in adrenal pheochromocytomas: clinical, histopathologic, and cell cycle/apoptosis gene expression analysis. Surgery 2008, 143(6): 759-768.
- Boltze C, Mundschenk J, Unger N, Schneider-Stock R, Peters B, Mawrin C, Hoang-Vu C, Roessner A, & Lehnert H: Expression Profile of the Telomeric Complex Discriminates between Benign and Malignant Pheochromocytoma. J Clin Endocrinol Metab 2003, 88(9): 4280-4286.
- Liu TH, Chen YJ, Wu SF, Gao J, Jiang WJ, Lu ZH, Guan J, Wei SZ, Luo YF, Cao JL, Wan JW: Distinction between benign and malignant pheochromocytomas. Zhonghua Bing Li Xue Za Zhi. 2004, 33(3): 198-202.
- Häyry V, Salmenkivi K, Arola J, Heikkilä P, Haglund C, & Sariola H: High frequency of SNAIL-expressing cells confirms and predicts metastatic potential of phaeochromocytoma. Endocr Relat Cancer 2009, 16(4): 1211-1218.
- Engels BM, & Hutvagner G: Principles and effects of microRNA-mediated post-transcriptional gene regulation. Oncogene 2006, 25(46): 6163-6169.
- Meyer-Rochow GY, Jackson NE, Conaglen JV, Whittle DE, Kunnimalaiyaan M, Chen H, Westin G, Sandgren J, Stålberg P, Khanafshar E et al: MicroRNA profiling of benign and malignant pheochromocytomas identifies novel diagnostic and therapeutic targets. Endocr Relat Cancer 2010, 17(3): 835-846.
- Latif F, Tory K, Gnarra J, Yao M, Duh F, Orcutt M, Stackhouse T, Kuzmin I, Modi W, Geil L et al: Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260(5112): 1317-1320.
- Neumann H, Berger DP, Sigmund G, Blum U, Schmidt D, Parmer RJ, Volk B, & Kirste G: Pheochromocytomas, Multiple Endocrine Neoplasia Type 2, and von Hippel-Lindau Disease. N Engl J Med 1993, 329(21): 1531-1538.
- White R, & O’Connell P: Identification and characterization of the gene for neurofibromatosis type 1. Curr Opin Genet Dev 1991, 1(1): 15-19.
- Neumann HPH, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K et al: Germ-Line Mutations in Nonsyndromic Pheochromocytoma. NEJM 2002, 346(19): 1459-1466.
- Baysal BE: Mutations in SDHD: a Mitochondrial Complex II Gene, in Hereditary Paraganglioma. Science 2000, 287(5454): 848-851.
- Hao H. X, Khalimonchuk O, Schraders M, Dephoure N, Bayley J. P, Kunst H, Devilee P, Cremers C. W. R. J, Schiffman JD, Bentz et al: SDH5, a Gene Required for Flavination of Succinate Dehydrogenase, Is Mutated in Paraganglioma. Science 2009, 325(5944): 1139-1142.
- Qin Y, Yao L, King EE, Buddavarapu K, Lenci RE, Chocron ES, Lechleiter JD, Sass M, Aronin N, Schiavi F et al: Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet 2010, 42(3): 229-233.
- Comino-Méndez I, Gracia-Aznárez F. J, Schiavi F, Landa I, Leandro-García LJ, Letón R, Honrado E, Ramos-Medina R, Caronia D, Pita G et al: Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet 2011, 43(7): 663-667.
- Moog S, Goncalves J, Menara M, Monnot C, Germain S, Gimenez-Roqueplo A. P & Favier J: Reprogrammation de la matrice extra-cellulaire dans les phéochromocytomes et paragangliomes SDHB-dépendants. Ann D’Endocr 2020, 81(4): 172.
- Jimenez C, Rohren E, Habra MA, Rich T, Jimenez P, Ayala-Ramirez M, & Baudin E: Current and Future Treatments for Malignant Pheochromocytoma and Sympathetic Paraganglioma. Curr Oncol Rep 2013, 15(4): 356-371.
- Janssen I, Blanchet EM, Adams K, Chen CC, Millo CM, Herscovitch P, Taieb D, Kebebew E, Lehnert H, Fojo AT, & Pacak K: Superiority of [68Ga]-DOTATATE PET/CT to Other Functional Imaging Modalities in the Localization of SDHB-Associated Metastatic Pheochromocytoma and Paraganglioma. Clin Cancer Res 2015, 21(17): 3888-3895.
- Banerji U: Heat Shock Protein 90 as a Drug Target: Some Like It Hot. Clin Cancer Res 2009, 15(1): 9-14.
- Powers MV, & Workman P: Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr Relat Cancer 2006: S125-S135.
- Semenza GL: Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov Today 2007, 12(19-20): 853-859.
- Yao JC, Phan AT, Chang DZ, Wolff RA, Hess K, Gupta S, Jacobs C, Mares JE, Landgraf AN, Rashid et al: Efficacy of RAD001 (Everolimus) and Octreotide LAR in Advanced Low- to Intermediate-Grade Neuroendocrine Tumors: Results of a Phase II Study. J Clin Oncol 2008, 26(26): 4311-4318.
- Druce MR, Kaltsas GA, Fraenkel M, Gross DJ, & Grossman AB: Novel and Evolving Therapies in the Treatment of Malignant Phaeochromocytoma: Experience with the mTOR Inhibitor Everolimus (RAD001). Horm Metab Res 2009, 41(09): 697-702.
- Nölting S, & Grossman AB: Signaling Pathways in Pheochromocytomas and Paragangliomas: Prospects for Future Therapies. Endocr Pathol 2012, 23(1): 21-33.
- Salmenkivi K, Heikkil P, Liu J, Haglund C, & Arola J: VEGF in 105 pheochromocytomas: enhanced expression correlates with malignant outcome. APMIS, 111(4) 2003: 458-464.
- Jimenez C, Cabanillas M. E, Santarpia L, Jonasch E, Kyle K. L, Lano E. A, Matin S. F, Nunez R. F, Perrier N. D, Phan A et al: Use of the Tyrosine Kinase Inhibitor Sunitinib in a Patient with von Hippel-Lindau Disease: Targeting Angiogenic Factors in Pheochromocytoma and Other von Hippel-Lindau Disease-Related Tumors. J Clin Endocrinol Metab 2009, 94(2): 386-391.
- Gross DJ, Munter G, Bitan M, Siegal T, Gabizon A, Weitzen R, Merimsky O, Ackerstein A, Salmon A, Sella A et al: The role of imatinib mesylate (Glivec) for treatment of patients with malignant endocrine tumors positive for c-kit or PDGF-R. Endocr Relat Cancer 2006, 13(2): 535-540.
- Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P, Munshi N, Anaissie E, Wilson C, Dhodapkar et al: Antitumor Activity of Thalidomide in Refractory Multiple Myeloma. NEJM 1999, 341(21): 1565-1571.
- Kulke MH, Stuart K, Enzinger PC, Ryan DP, Clark JW, Muzikansky A, Vincitore M, Michelini A, & Fuchs CS: Phase II Study of Temozolomide and Thalidomide in Patients With Metastatic Neuroendocrine Tumors. J Clin Oncol 2006, 24(3): 401-406.
- Choi HJ, Song BJ, Gong YD, Gwak WJ, & Soh Y: Rapid degradation of hypoxia-inducible factor-1α by KRH102053, a new activator of prolyl hydroxylase 2. Br J Pharmacol 2008, 154(1): 114-125.
- Bruchelt G, Klingebiel T, Treuner J, Beck J, Lode H, Seitz G & Niethammer D: Radiolabeled Metaiodobenzylguanidine (Mibg) In Diagnosis And Therapy Of Neuroblastoma - Results From Basic Research (Review). Int J Oncol 1995.
- Bhatia KSS, Ismail MM, Sahdev A, Rockall AG, Hogarth K, Canizales A, Avril N, Monson JP, Grossman AB, & Reznek RH: 123I-metaiodobenzylguanidine (MIBG) scintigraphy for the detection of adrenal and extra-adrenal phaeochromocytomas: CT and MRI correlation. Clin Endocrinol (Oxf) 2008, 69(2): 181-188.
- van Hulsteijn LT, Niemeijer ND, Dekkers OM, & Corssmit EPM: 131I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: systematic review and meta-analysis. Clin Endocrinol (Oxf) 2013, 80(4): 487-501.
- Parenti G, Zampetti B, Rapizzi E, Ercolino T, Giachè V & Mannelli M: Updated and New Perspectives on Diagnosis, Prognosis, and Therapy of Malignant Pheochromocytoma/Paraganglioma. J Oncol 2012.
- Yu L, Fleckman AM, Chadha M, Sacks E, Levetan C, & Vikram B: Radiation Therapy of Metastatic Pheochromocytoma: Case Report and Review of the Literature. Am J Clin Oncol 1996, 19(4): 389-393.
- Collins D: Bailey & Love’s Short Practice of Surgery. 25th Edn N. S. Williams, C. J. K. Bulstrode and P. R. O’Connell (eds) 283 × 225 mm. Pp. 1514. Illustrated. 2008. Hodder Arnold: London. Br J Surg 2008, 95(10): 1311.
Annals of urologic oncology
p-ISSN: 2617-7765, e-ISSN: 2617-7773
 Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.