
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
Vascular Neoplasms and Related Lesions of Kidney
Michelle S. Lin1, Jae Y. Ro1
1Department of Pathology and Genomic Medicine, Houston Methodist Hospital, Weill Cornell Medical College, Houston, TX 77030, USA.
Correspondence: Jae Y. Ro(Department of Pathology and Genomic Medicine, Houston Methodist Hospital, Weill Cornell Medical College, Houston, TX 77030, USA; Email: jaero@houstonmethodist.org).
Annals of Urologic Oncology 2020, 3(1): 1-14. https://doi.org/10.32948/auo.2020.05.02
Received: 16 Feb 2020 | Accepted: 27 April 2020 | Published online: 25 Jun 2020
Vascular tumors arising in the kidney are distinctly uncommon occurrences and have not been well-characterized in the literature; most cases have been documented in single reports or small case series. These tumors often exhibit nonspecific clinical and radiologic features and may mimic malignant epithelial tumors such as renal cell carcinoma (RCC) on imaging studies, necessitating nephrectomy with pathologic examination for definitive diagnosis. In addition, since primary renal vascular tumors are rare and infrequently encountered in surgical pathology specimens, they are often not considered in the differential diagnosis and therefore may be systematically under-diagnosed. In this review, we discuss the clinical and morphologic features of benign, borderline, and malignant vascular neoplasms and related lesions arising in the kidney, with a focus on recent, clinically relevant advances in imaging, histopathologic diagnosis, and treatment.
Key words vascular tumors; Hemangioma; Anastomosing hemangioma; Angiosarcoma; Kidney
Vascular tumors of the kidney are relatively rare, and their clinicopathologic characteristics have not been well delineated. Due to their uncommonness, as well as their nonspecific clinical presentation and imaging findings often leading to the impression of more common malignant epithelial tumors such as renal cell carcinoma (RCC), diagnosis can pose a challenge for radiologists and pathologists alike. In addition, the description by Montgomery and Epstein in 2009 of anastomosing hemangioma, a benign vascular neoplasm arising most often in the kidney and showing overlapping features with angiosarcoma, has added further difficulties in the histopathologic differential diagnosis of renal vascular tumors [1]. In this review, we discuss the clinical, radiographic, and histopathologic features of a wide range of vascular neoplasms that arise from the kidney (Table 1), including benign tumors (hemangioma and anastomosing hemangioma), borderline tumors (epithelioid and other hemangioendotheliomas), malignant tumors (angiosarcoma), tumor-like lesions (arteriovenous malformation, lymphangioma, and intravascular papillary endothelial hyperplasia), and various other vascular-related tumors (hemangioblastoma, juxtaglomerular cell tumor, glomus tumor, and myopericytoma).
|
Table 1. Summary of vascular lesions of kidney. |
||||
|
|
Clinical Features |
Radiologic Features |
Histologic Features |
Prognosis/Treatment |
|
Benign vascular tumors
|
|
|
|
|
|
Hemangioma |
Age: peak 30-40 years Size: most < 1-2 cm Location: tip of papilla most common |
US: variably echogenic CT: hypodense to isodense, variable enhancement MRI: T1 hypointense, T2 hyperintense
|
Cavernous: composed of dilated vascular channels Capillary: composed of capillary-sized channels
|
Observation (if asymptomatic) Endoscopic ablation, surgical resection
|
|
Anastomosing hemangioma |
Age: median 49 years Size: mean 1.5 cm Associations: ESRD
|
US: variably echogenic CT: hypodense to isodense, heterogeneous enhancement MRI: T2 hyperintense
|
Anastomosing capillary-sized vascular channels lined by hobnailed cells Fibrin thrombi, EMH, hyaline globules may be seen |
Prognosis: excellent Treatment: nephrectomy (due to pre-operative suspicion for malignancy)
|
|
Borderline and malignant vascular tumors
|
|
|
|
|
|
Epithelioid hemangioendothelioma |
Age: median 53 years Size: median 3.8 cm
|
US: hypoechoic CT: no significant enhancement MRI: heterogeneous T1, hypointense T2, heterogeneous enhancement
|
Cords/nests of epithelioid cells in myxohyaline stroma Intracytoplasmic lumina containing RBCs may be seen |
Prognosis: not established, has potential to metastasize Treatment: nephrectomy |
|
Angiosarcoma
|
Age: 6th-7th decades Sex: M:F of 7:1 Size: mean 13 cm |
US: solid hypoechoic mass CT: hypodense, variable enhancement MRI: intermediate to hyperintense T1, “striated” T2 signal |
Architecture ranges from well-formed sinusoidal vascular channels to solid sheets of cells Varying amounts of multilayering, cytologic atypia, mitoses, necrosis |
Prognosis: poor, mean survival 7 months Treatment: nephrectomy with adjuvant chemo/ radiation (not well-established) |
|
Vascular tumor-like lesions
|
|
|
|
|
|
Arteriovenous malformation |
Age: 3rd-4th decades Presentation: may lead to HTN, HF
|
US: hypoechoic cystic structures, high-velocity flow on Doppler CT: masses of vascular density MRI: T2 signal voids, early enhancement
|
Collection of tortuous thick and thin-walled vessels
|
Embolization preferred treatment Surgical resection if embolization not able to be performed
|
|
Lymphangioma |
Age: diagnosed during childhood or over age 40 Location: peripelvic region or renal sinus, may be bilateral |
US: hypoechoic cystic lesion CT: hypodense, possible peripheral enhancement MRI: T1 hypointense, T2 hyperintense
|
Aggregation of thin-walled cystic spaces lined by endothelial cells Positive for lymphatic markers (D2-40) |
Observation (if asymptomatic) Aspiration, marsupialization, sclerotherapy, nephrectomy |
|
Intravascular papillary endothelial hyperplasia
|
Age: mean 49 years Sex: M:F of 3.3:1 Size: mean 3 cm Location: mid-portion or hilar region |
US: variably echogenic CT: isodense, intense peripheral enhancement MRI: T1 hypo/isointense, T2 hyperintense |
Intravascular papillary proliferation Papillae have hyalinized fibrinous cores and are lined by bland endothelial cells
|
Prognosis: excellent Treatment: nephrectomy due to pre-operative suspicion of malignancy |
|
Other vascular-related tumors
|
|
|
|
|
|
Hemangioblastoma |
Age: mean 47 years Size: mean 4.1 cm |
CT: hypodense, heterogeneous enhancement
|
Sheets of polygonal cells with vacuolated cytoplasm, with complex vascular network Positive for inhibin, NSE, S100
|
Prognosis: excellent Treatment: nephrectomy (due to pre-operative impression of RCC) |
|
Glomus tumor |
Age: mean 53 years Size: mean 4.2 cm |
US: hyperechoic, heterogeneous mass CT: hypodense, moderate enhancement |
Solid sheets of monotonous round cells within vascular network Positive for SMA, calponin, caldesmon, muscle-specific actin
|
Prognosis: typically good; cases with atypical features can metastasize Treatment: nephrectomy |
|
Myopericytoma |
Age: mean 49 years Size: mean 5.6 cm
|
US: hypoechoic, heterogeneous mass CT: hypodense, heterogeneous enhancement |
Concentric proliferation of spindled cells around slit-like thin-walled vascular spaces Positive for SMA, caldesmon, muscle-specific actin
|
Prognosis: good Treatment: nephrectomy |
|
Juxtaglomerular cell tumor |
Age: peak 2nd-3rd decades Sex: M:F of 1:2 Size: most < 3 cm Presentation: HTN and hypokalemia |
US: hypoechoic CT: isodense or hypodense, no significant enhancement MRI: T1 isointense, T2 variably hyperintense, mild enhancement |
Sheets of uniform polygonal cells within vascular network Positive for CD34, CD117, SMA, desmin, caldesmon, renin Rhomboid-shaped crystals on EM
|
Prognosis: typically good, rare cases of metastasis Treatment: tumor resection |
|
US - ultrasound; CT - computed tomography; MRI - magnetic resonance imaging; ESRD - end-stage renal disease; HTN - hypertension; HF - heart failure; NSE - neuron-specific enolase; RBC - red blood cell; RCC - renal cell carcinoma; SMA - smooth muscle actin.
|
||||
Hemangioma
Hemangiomas are the most common vascular neoplasms of the kidney, with over 200 cases documented in the literature to date [2]. While its incidence in the kidney is still extremely low as compared to epithelial tumors, the kidney is the second most common location of visceral-based hemangiomas, following the liver [3]. These tumors are most often diagnosed in young to middle-aged adults, particularly those in the age group of 30-40 years; no particular sex predilection has been observed [3, 4]. Many renal hemangiomas do not produce any symptoms and are only detected incidentally during workup for unrelated conditions; the most commonly reported symptoms are hematuria and abdominal or flank pain [2, 4]. Occasionally, renal hemangiomas are seen in the context of end-stage renal disease, polycythemia, or hypertension [5-9]. Rare cases arising in the vicinity of RCCs have been reported, as well as cases with syndromic associations such as Klippel-Trenaunay and Sturge-Weber disease [8, 10-14].
Most hemangiomas are small lesions measuring less than 1-2 cm, but lesions up to 30 cm have been reported [15, 16]. They are most frequently located within the renal pelvis or medulla, particularly the tip of the papilla [17]. Imaging characteristics are nonspecific, with ultrasound studies showing variably echogenic masses and computed tomography (CT) typically demonstrating hypodense or isodense masses with widely differing degrees of contrast enhancement, ranging from no enhancement, to irregular enhancement, to intense enhancement persisting to the venous phase [4, 11, 17-19]. Some cases have demonstrated central anechoic areas on ultrasound or central hypo-attenuation on CT, corresponding to blood-filled spaces in the center of the lesion [5, 18]. Magnetic resonance imaging (MRI) generally shows hypointensity on T1-weighted images and marked hyperintensity on T2-weighted images [4, 17]. Overall, the radiologic features are often indistinguishable from malignancies such as RCC and urothelial carcinoma (which also typically presents in the renal pelvis) [18, 20]. Some studies have suggested that renal hemangiomas possess similar imaging findings to their hepatic counterparts (especially on dynamic phase CT and MR imaging) which may assist in their recognition as vascular neoplasms, but others have observed less avid enhancement compared to hemangiomas in the liver, perhaps secondary to prior intratumoral hemorrhage [17, 21].
Grossly, hemangiomas appear as well-demarcated, unencapsulated hemorrhagic masses with a spongy tan-red cut surface [15]. Microscopically, hemangiomas may be of the cavernous or capillary subtype, with the cavernous subtype more frequently encountered [2, 4]. Cavernous hemangiomas are characterized by dilated vascular channels containing erythrocytes, while capillary hemangiomas show a proliferation of capillary-sized blood vessels separated by scant connective tissue stroma [2, 9, 22]. Interestingly, unlike capillary hemangiomas arising in the skin and soft tissue, a distinct lobular architectural growth pattern is not seen in renal capillary hemangiomas [2]. In both subtypes, the vascular spaces are lined by flattened, bland endothelial cells without evidence of nuclear atypia or mitosis [9, 15]. On immunohistochemistry, the cells show reactivity for vascular markers, including CD34, CD31, FVIII, ERG, and FLI-1.
The histopathologic differential diagnosis of renal hemangioma includes conventional clear cell RCC (ccRCC), which is often the working clinical and radiologic diagnosis. While ccRCC typically shows very distinctive morphology, infrequently, cases with degenerative changes may show attenuation of the epithelial component and subsequent accentuation of the rich vascular network, leading to a resemblance to hemangioma [15, 23]. In addition, a new subtype of RCC termed “hemangioma-like RCC” has recently been described and should also be considered in the differential diagnosis [23-25]. Immunohistochemical analysis using cytokeratins, PAX8, CD10, and CAIX should resolve the distinction between RCC and hemangioma. Other neoplasms that should be ruled out include angiosarcoma (which typically shows increased cytologic atypia, mitotic activity, and necrosis), anastomosing hemangioma (which usually displays lobular growth of sinusoidal vascular spaces with prominent intervening stroma), and angiomyolipoma (especially as entrapped adipocytes may be seen in hemangiomas, producing an angiomyolipoma-like appearance; immunohistochemistry with melanocytic markers usually easily distinguishes these two entities) [12, 15].
The approach to management of renal hemangiomas depends on several clinical factors, including the size and location of the tumor, the patient’s symptoms, and the ability to render a conclusive pre-operative diagnosis (which sometimes can be done by visualizing the lesion via transurethral ureterorenoscopy) [4, 22, 26]. If a pre-operative diagnosis of hemangioma can be confirmed, then observation is acceptable for asymptomatic patients, and endoscopic ablation can be performed for symptomatic patients with accessible lesions near the renal pelvis [2, 26-27]. Surgical resection is performed for patients with significant symptoms (such as hemorrhage or spontaneous rupture) or with presentation and radiologic findings suggestive of malignancy [2, 18]. Nephron-sparing partial nephrectomy can be performed for small hemangiomas, while total nephrectomy may be required for larger hemangiomas [27, 28].
Anastomosing hemangioma
Anastomosing hemangioma (AH) is a distinctive variant of hemangioma, first described by Montgomery and Epstein in 2009; it is characterized by sinusoidal anastomosing architecture and thus may mimic a low-grade angiosarcoma [1, 29]. To date, over 60 cases of AH in the kidney have been reported, as well as a similar number of extrarenal cases arising from a wide variety of sites including the testis, ovary, adrenal gland, liver, colon, small bowel, soft tissue, bone, and breast [30, 31]. The median age of occurrence is 49 years for AH arising from the kidney and 65 years for AH arising from extra-renal sites [32]. They are generally small tumors (mean 1.5 cm) located within the renal parenchyma, most frequently the medulla [32]. In the majority of cases, AH produces no symptoms and is incidentally detected, but symptomatic cases have reported hematuria, back pain, and lower urinary tract symptoms [29, 30]. Studies have found that approximately two-thirds of renal AH are associated with end-stage renal disease (ESRD); these tumors tend to be smaller, are more often multifocal, and arise in younger patients [30, 32-35]. In addition, 23% of renal AH are seen in conjunction with a different renal neoplasm, including ccRCC, papillary RCC, and metanephric adenoma; in many cases detection of these tumors led to the incidental discovery of AH [32, 34].
The radiologic features of AH are nonspecific and may be virtually impossible to distinguish from malignant tumors of the kidney, including RCC and angiosarcoma [20, 30, 33]. Ultrasonography demonstrates variably echogenic lesions, while CT generally shows lobulated, hypodense to isodense solid masses with progressive heterogeneous contrast enhancement persisting to the venous phase [20, 29, 36, 37]. Some cases demonstrate avid peripheral nodular enhancement highly suggestive of RCC [20]. Features of AH on MRI are not well-established, but some studies report hyperintensity on T2-weighted images with variable enhancement [38]. Overall, as the radiologic appearance may mimic malignancies, particularly hypervascular RCC, pre-operative diagnosis of AH may not be possible on imaging. This consideration may carry significant implications, especially for ESRD patients awaiting a renal transplant for whom active malignancy is a contraindication to transplant [20, 33].
On gross examination, AH typically appears as a well-demarcated but unencapsulated mass with a tan-mahogany, hemorrhagic, and spongy cut surface [34]. Microscopically, AH may display a diffuse or vaguely lobulated architecture and is composed of a proliferation of capillary-sized anastomosing vascular channels that may bear a superficial resemblance to splenic sinusoids [1, 29]. The vascular spaces are lined by bland, uniform endothelial cells, sometimes with a focal hobnailed appearance, and are set within an abundant hyalinized stroma [1, 29, 30]. Intravascular fibrin thrombi are frequently seen, as are foci of extramedullary hematopoiesis (more commonly encountered in cases of AH associated with ESRD) and PAS-positive eosinophilic hyaline globules which resemble those seen in Kaposi’s sarcoma [1, 30, 32]. Immunohistochemical studies show reactivity to vascular markers and a low Ki-67 proliferation index, as well as negativity for GLUT-1 (a marker for capillary hemangioma), CD8 (a marker for splenic sinusoids), and D2-40 [2, 30]. A recent study of AH has revealed recurrent GNAQ mutations, which have also been found in other benign vascular tumors such as capillary hemangiomas but not in angiosarcomas [30, 39].
The most important entity to exclude in the differential diagnosis is a low-grade angiosarcoma, which also shows prominent anastomosing architecture. In contrast to AH, angiosarcomas are typically larger lesions which display broadly infiltrative growth, multilayering or tufting of the endothelium, and variable degrees of necrosis, cytologic atypia, and mitotic activity [30, 33]. Masson tumor (intravascular papillary endothelial hyperplasia) may display complex papillary architecture which may fuse and resemble anastomosing channels, but the predominantly intravascular growth and the distinctive morphology of hyalinized papillary cores lined by hyperplastic endothelial cells can help facilitate its distinction from AH [30, 36]. Finally, as with conventional hemangioma, entities such as vascular-predominant angiomyolipoma and ccRCC with degenerative changes of the neoplastic cells (thus leaving behind the complex vascular network) should also be considered and excluded during the diagnostic workup [29, 30, 33].
AH in the kidney has an excellent overall prognosis, with no reports of local recurrence or metastasis to date [30]. Most patients are treated with nephrectomy due to suspicion for malignancy on clinical and radiologic workup. However, it is possible that better characterization of specific features of AH on imaging in combination with increased utilization of core needle biopsies may spare patients unnecessary nephrectomies and the accompanying morbidities in the future [20, 30].
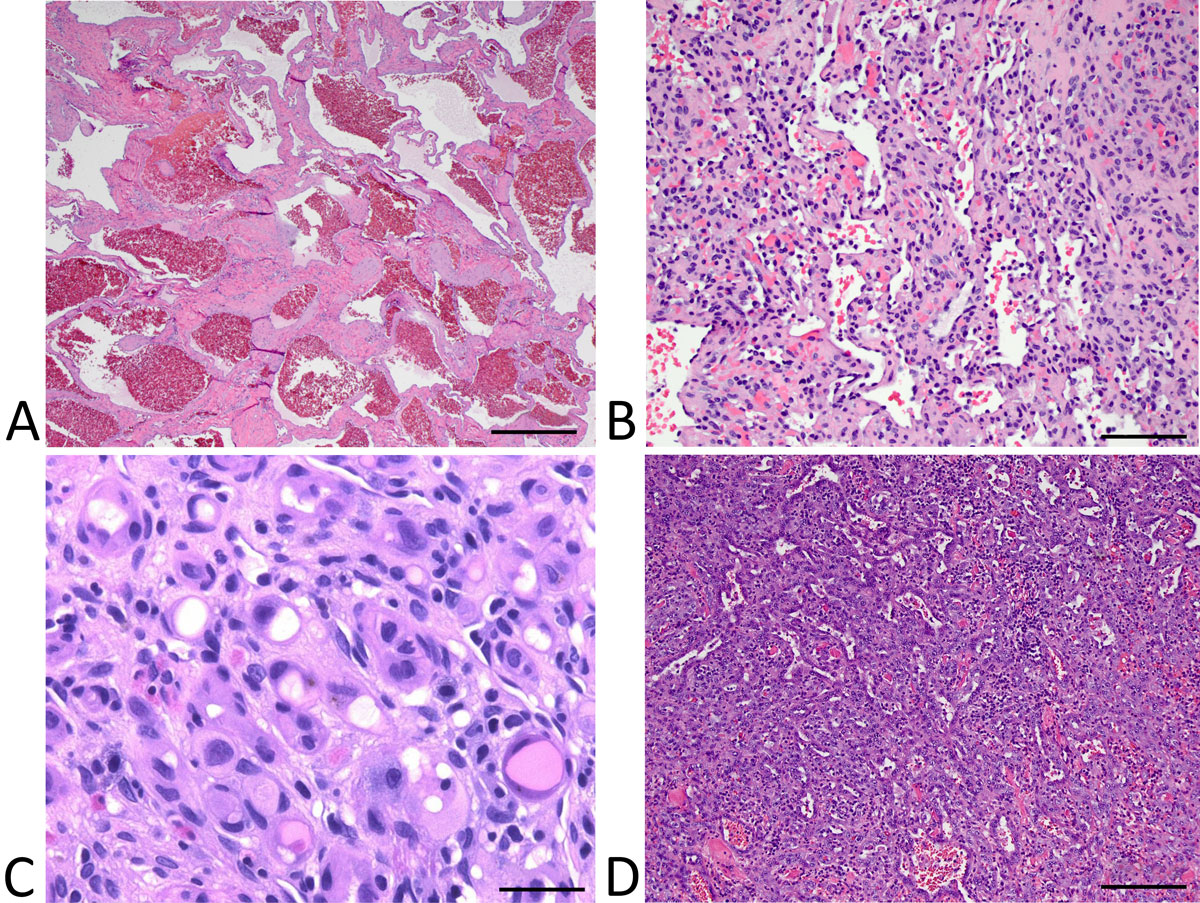
Figure 1. (A) Cavernous hemangioma exhibiting a thick-walled dilated vascular channels containing erythrocytes; (B) anastomosing
hemangioma composed of anastomosing vascular spaces lined by bland, focally hobnailed endothelial cells; (C) epithelioid
hemangioendothelioma demonstrating epithelioid cells with intracytoplasmic lumina within a sclerotic stroma; and (D) angiosarcoma showing
irregularly interanastomosing vascular channels lined by pleomorphic, focally stratified endothelial cells. Scale bar: (a)=500 μm, (b)=250 μm,
(c)=100 μm, (d)=250 μm.
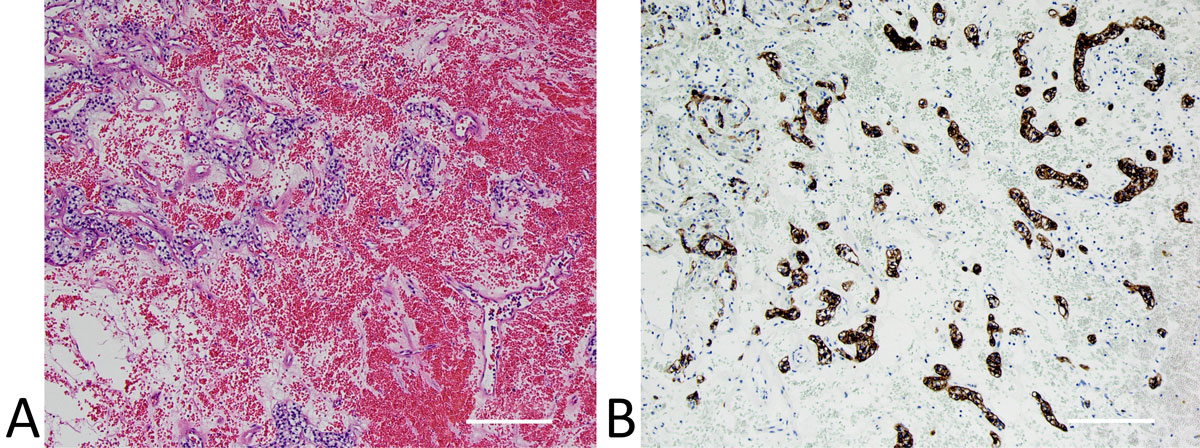
Figure 2. (A) Hemangioma-like clear cell renal cell carcinoma demonstrating a prominent vascular component and thus bearing strong resemblance to hemangioma; (B) immunostaining for pan-cytokeratin highlighting the neoplastic epithelial cells. Scale bar: (a)=250 μm, (b)=250
Epithelioid hemangioendothelioma
Epithelioid hemangioendothelioma (EHE) is a vascular tumor with generally intermediate biologic behavior but notably harboring the potential to metastasize [40]. While EHE can occur at a variety of locations (most commonly soft tissue, lung, and liver), primary EHE arising from the kidney is exceedingly rare with only five cases reported in the literature to date [41-45]. The median age of occurrence is 53 years and no particular sex predilection has been observed. Patients may present with incidentally detected lesions, hematuria, or abdominal pain. Most reported cases are under 4 cm in size (median size 3.8 cm), but one case with a large 14-cm lesion has been documented [43].
The radiologic features of primary renal EHE have not been well-characterized due to the paucity of reported cases. Single-case studies have demonstrated hypoechogenicity on ultrasound examination and solid lesions lacking significant enhancement on CT imaging [43, 45]. On MRI, Xiao et al. reported heterogeneous intensity on T1-weighted images with diminished intensity on T2-weighted images and heterogeneous, persistent enhancement with contrast administration [44]. Overall, the clinical and radiologic findings are nonspecific and preclude definitive pre-operative diagnosis.
On gross examination, EHE is a well-demarcated, variably nodular or lobulated tumor with a firm gray-red to gray-white cut surface [41, 44]. Microscopically, a proliferation of epithelioid cells forming cords, nests, and slit-like spaces within a characteristically abundant sclerotic or myxohyaline stroma is seen [46]. The cells occasionally display intracytoplasmic lumina containing erythrocytes. In most cases no necrosis or mitosis is seen; one case of metastatic EHE did show extensive areas of necrosis and numerous mitoses [43, 46]. Immunohistochemically, the cells of EHE generally stain positively for vascular markers and negatively for epithelial markers; some cases show reactivity for smooth muscle actin (SMA) as well [41, 43].
Due to its epithelioid morphology, EHE can present a diagnostic dilemma, especially in unusual sites such as the kidney. The differential diagnosis is wide and includes metastatic carcinoma and melanoma, which usually can be resolved with immunohistochemical staining (however, focal cytokeratin positivity is not uncommonly seen in soft tissue EHE and is a pitfall to making this diagnosis) [45]. Epithelioid angiosarcoma may also be considered in the differential; it can be distinguished from EHE by the presence of poorly formed anastomosing vascular spaces, as well as necrosis and increased mitotic activity [45]. Finally, epithelioid angiomyolipoma may also demonstrate a superficial resemblance to EHE, but it typically displays alveolar or sheet-like growth with nests of cells separated by vascular septa, and also characteristically shows strong positivity for melanocytic markers such as HMB-45 and Melan-A [45].
The prognosis and standard treatment of primary renal EHE is unclear due to the scarcity of documented cases. Most cases were treated with partial or total nephrectomy and had no evidence of disease with up to 6 months follow-up. One case reported metastasis to the liver, paracaval lymph nodes, and mesocolon shortly following surgical resection [43]. The patient was treated successfully using sunitinib (a receptor tyrosine kinase with anti-angiogenic activity), with no progression of disease 33 months post-operatively. As the disease-related mortality of EHE varies tremendously based on the primary site – from 13% in soft tissue to 35% in the liver and 65% in the lung – more experience with EHE arising from the kidney is needed to better characterize its overall behavior and clinical course [46].
Other hemangioendotheliomas
In addition to the five cases of EHE discussed above, one case of kaposiform hemangioendothelioma (KHE) and one case of composite hemangioendothelioma (CHE) occurring in the kidney have also been reported in the literature [47, 48]. KHE is considered a borderline vascular tumor which may be locally aggressive but typically does not metastasize. It usually occurs during early childhood in cutaneous or retroperitoneal sites and may be associated with the Kasabach-Merritt phenomenon [47]. The single case of KHE arising in the kidney was described in a 4-year-old female who presented with hematuria and was found to have a 13-cm mass in the left kidney with extension to the inferior vena cava as well as involvement of the right atrium and lymph nodes [47]. The mass was initially thought to represent Wilms’ tumor but showed little response to chemotherapy, so radical nephrectomy was subsequently performed. Histopathologic examination showed capillary hemangioma-like areas and Kaposi sarcoma-like areas containing fascicles and glomeruloid structures composed of spindled endothelial cells; numerous mitoses with atypical mitotic figures were also present. The patient was treated with interferon-alpha-2b (an anti-angiogenic factor) and was free of disease with eight years post-operative follow-up [47].
CHE is a recently described borderline vascular neoplasm which exhibits varying components of different benign, intermediate, and malignant vascular tumors [48]. It is exceedingly rare, with less than 40 cases reported in the literature, mainly in the cutaneous extremities [48]. Zhang et al. describe a case of CHE arising from the kidney in a 32-year-old female who presented with an enlarging 2.6-cm mass in the right kidney [48]. A percutaneous needle biopsy was initially performed and the findings were interpreted as “atypical vascular neoplasm.” Microscopic exam of the subsequent total nephrectomy specimen showed an infiltrative lesion with an admixture of different histologic patterns including areas resembling angiosarcoma, EHE, and spindle cell hemangioma. No adjuvant treatment was administered and the patient remained free of disease 11 months post-operatively. Notably, CHE does possess the ability to metastasize, but no deaths from disease have been documented to date [48].
Angiosarcoma
Angiosarcoma is a malignant vascular neoplasm which may occur in any location of the body, including the skin, soft tissue, breast, bone and visceral organs including the liver. Primary renal angiosarcoma is very rare, with about 70 cases described in the literature to date [49-57]. Most of these cases are seen in older men, with a peak incidence in the sixth to seventh decades and a pronounced male predilection (male-to-female ratio of 7:1) [49]. These tumors tend to be relatively large, with a mean size of 13 cm, and thus are usually symptomatic at presentation, with the most commonly reported symptoms being flank pain, hematuria, and palpable mass, as well as non-specific systemic symptoms such as fever, fatigue, and weight loss [49]. No association with traditional risk factors of angiosarcoma, including vinyl chloride, thorotrast, arsenic, radiation, or chronic lymphedema, has been observed in renal cases [51].
Imaging features of renal angiosarcoma are variable, nonspecific, and generally indistinguishable from those of RCC. Ultrasound examination shows a large solid hypoechoic renal mass; one case exhibited peripheral “egg-shell” and central calcifications [58, 59].CT imaging demonstrates a hypodense mass with varying levels of contrast enhancement ranging from none, to heterogeneous, to intense peripheral, to diffuse enhancement [53, 54, 58, 59]. Central areas of necrosis or hemorrhage may be detected [55]. MRI studies have shown intermediate to high signal intensity on T1-weighted images, and some cases show alternating areas of high and low signal intensity on T2-weighted images, creating a striated pattern corresponding to alternating necrotic and solid areas of the tumor [59]. Some reports have also shown signal voids in the periphery on T2-weighted images, corresponding to the neoplastic vascular channels [54].
Grossly, renal angiosarcoma presents as a poorly demarcated hemorrhagic mass with a dark-red, spongy cut surface and variable amounts of necrosis [51]. On histopathologic exam, the architecture can range from well-formed anastomosing vascular channels to solid sheets of epithelioid or spindled cells lacking or with only subtle areas of vasoformation [47]. The neoplastic endothelial cells show areas of multilayering, intraluminal budding and tufting, as well as increased cytologic atypia and mitotic activity [49]. Generally, a diffusely infiltrative growth pattern is seen, with some cases displaying invasion of the perinephric adipose tissue [49]. On immunohistochemistry, the neoplastic cells should show positivity for at least one endothelial marker (CD34, CD31, ERG, FLI-1) and are negative for epithelial markers; however, cases of epithelioid angiosarcoma have been shown to stain positively for keratins [2, 49, 55]. The Ki-67 proliferative index is usually very high, ranging from 30% to over 80% [51].
The differential diagnosis of renal angiosarcoma includes metastatic angiosarcoma from another site, which occurs more commonly than primary renal angiosarcoma [15]. Knowledge of clinical history, as well as imaging features demonstrating a large renal mass with multiple smaller lesions in the liver or lung (favoring a primary renal lesion), can help to determine the site of origin [2, 49]. Anastomosing hemangioma is another important differential diagnostic consideration as it histologically mimics angiosarcoma; however it is typically smaller and lacks the diffuse infiltration, multilayering endothelium, coagulative necrosis, cytologic atypia, and increased mitotic activity seen in angiosarcoma [15, 49]. Finally, sarcomatoid RCC may resemble a poorly differentiated angiosarcoma and thus should be ruled out using immunostains for keratins, PAX8, CD10, and CAIX [2, 49].
The prognosis of primary renal angiosarcoma is grim, with metastases developing in nearly all patients and death from disease in the majority of cases at a mean of 7 months [49]. The most common sites of metastasis were lung and liver, but reports of metastasis to the bone, soft tissue, peritoneum, spleen, and lymph nodes have also been documented [49]. Important prognostic factors include the presence of metastatic disease at presentation and tumor size, with patients whose tumors were less than 5 cm generally demonstrating a higher survival rate [52, 57]. The management of renal angiosarcoma is controversial and not well-established; most patients were treated with radical nephrectomy followed by varying combinations of radiation and chemotherapy [51]. One recent study describes a case treated with neoadjuvant chemoradiation with excellent response, demonstrating potential for this therapeutic approach in future cases [56]. While targeted treatment with anti-angiogenic factors has been utilized with some success in non-renal angiosarcomas, two cases of renal angiosarcoma treated with recombinant interleukin-2 and bevacizumab ultimately resulted in death from disease [49, 58]. Nevertheless, experience remains limited and the role of adjuvant therapies in the treatment of renal angiosarcoma remains to be further elucidated.
Arteriovenous malformation
Arteriovenous malformations (AVMs) are characterized by an abnormal communication between the arterial and venous systems and may be either acquired or congenital [2]. Acquired AVMs, also known as arteriovenous fistulas (AVFs), are most often caused by trauma or iatrogenic injuries such as surgery or biopsy [2, 9]. Congenital AVMs, the focus of this discussion, are much less common than AVFs, with an overall prevalence of less than 1% in the general population [60]. AVMs are usually asymptomatic until around the third or fourth decade of life, when they may produce symptoms such as hematuria, flank pain, hypertension, or high-output heart failure [60]. A few studies have observed AVMs arising adjacent to hemangiomas and RCCs, possibly secondary to the production of angiogenic factors from these lesions [9].
On ultrasonography, AVMs appear as an area of hypoechoic irregular interconnecting cystic structures, with high-velocity turbid flow on Doppler [61]. CT studies show round or oval masses of vascular density which may show early enhancement of the renal vein and inferior vena cava [60, 61]. In some cases, the CT findings may mimic malignancy such as RCC, urothelial carcinoma, or lymphoma [9]. CT angiography can facilitate visualization of the abnormal tortuous vessels characteristic of AVM [61]. On MRI, numerous convoluted signal voids on T2-weighted images corresponding to the vessels can be seen, with enhancement in the early arterial phase [61, 62]. Catheter angiography is the gold standard for the diagnosis and evaluation of AVMs as it can most accurately demonstrate the vascular anatomy of the lesion, including its arterial supply and venous drainage [62, 63].
AVMs are not commonly encountered as surgical pathology specimens, as they are normally able to be successfully diagnosed radiologically and treated by embolization. The microscopic appearance of renal AVMs is identical to those elsewhere in the body, characterized by abnormally arranged, tortuous thick and thin-walled vessels with no intervening cellular elements, and occasionally with thrombosis [2, 9]. While the histopathologic diagnosis of AVM is usually relatively straightforward, entities which may potentially mimic AVM include angiomyolipoma and hemangioma [9]. Angiomyolipoma also demonstrates abnormal thick-walled vessels (and AVMs may entrap adipose tissue as well); however, it also shows the characteristic radiation of smooth muscle cells from the vessel walls as well as reactivity to melanocytic markers. Hemangioma is usually composed of uniform dilated or capillary-sized vessels, in contrast to the irregular tortuous vessels seen in AVM.
Endovascular therapy with selective embolization is the preferred treatment for AVM, as it is minimally invasive and nephron-sparing [60, 63]. Surgical therapy with partial or total nephrectomy is performed for unstable patients or patients with anatomically complex AVMs not amenable to embolization [60].
Lymphangioma
Lymphangiomas are benign malformations of the lymphatic system, hypothesized by some to represent a congenital developmental anomaly rather than a true neoplastic process [4]. Most commonly occurring in the neck or axilla, lymphangiomas arising from the kidney are very rare, with about 50 cases reported in the literature [64]. Most cases are diagnosed during childhood or over the age of 40; no particular sex predilection has been observed [4, 65]. Patients may present with flank pain, abdominal mass or hematuria; however, the majority of cases are asymptomatic and discovered incidentally during imaging workup for unrelated causes [4].
Renal lymphangiomas are most often located in the peripelvic region or renal sinus, and not infrequently involve the bilateral kidneys [4]. Ultrasound examination typically shows a hypoechoic cystic lesion, while CT shows a unilocular or multilocular hypodense cystic mass which may demonstrate peripheral or septal enhancement [65-68]. On MRI, lymphangiomas usually appear hypointense on T1-weighted imaging and hyperintense on T2-weighted imaging with varying degrees of enhancement [66, 67]. The radiologic differential diagnosis includes multilocular cystic RCC (which often shows intense septal enhancement or a solid enhancing nodule) as well as localized cystic disease of the kidney and multilocular cystic nephroma [67, 68].
Macroscopically, lymphangioma appears as a well-encapsulated multiloculated cystic mass containing clear fluid [4, 15]. Histopathologic exam demonstrates numerous thin-walled cystic spaces filled with proteinaceous fluid and lined by flattened bland endothelial cells [9, 15]. These cells stain positively for vascular markers, such as CD31 and Factor VIII, as well as lymphatic markers such as D2-40 [9, 15]. In the kidney, the main differential diagnosis of lymphangioma is with cystic nephroma, which can be distinguished from the former by the presence of ovarian-type stroma and the lack of reactivity to vascular and lymphatic markers on immunohistochemical studies [15].
Management of renal lymphangioma is dependent on the patient’s symptoms and the ability to render a pre-operative diagnosis on imaging studies. Small, asymptomatic lesions do not require treatment and may be observed [4]. If the diagnosis of lymphangioma can be confirmed radiologically, non-surgical methods such as percutaneous or laparoscopic aspiration, marsupialization, or sclerotherapy may be considered [65, 67]. Nephrectomy is reserved for more complicated cases and for cases in which lymphangioma was not recognized pre-operatively [66].
Intravascular papillary endothelial hyperplasia
Intravascular papillary endothelial hyperplasia (IPEH), also known as Masson’s tumor, is a reactive intravascular proliferation associated with stasis and thrombosis, and which may arise from dilated vessels, pre-existing vascular lesions, or hematomas [9]. IPEH mainly occurs in the skin and soft tissue; solid organ involvement is rare, and only 13 cases of IPEH arising from the kidney have been described in the literature to date [69]. The mean age of occurrence is 49 years, with a male predominance of cases (male-to-female ratio 3.3:1). Cases presented incidentally or with nonspecific symptoms such as flank pain or hematuria [69]. Most lesions were relatively small, with a mean size of 3 cm, and located in the mid-portion or hilar region of the kidney, arising from the renal vein, renal sinus, or vessels in the renal parenchyma [69].
Ultrasound examination of IPEH shows variable echogenicity, and CT demonstrates a heterogeneous mass isodense to the renal parenchyma, frequently with intense peripheral enhancement [69, 70]. On MRI, the lesion is hypointense or isointense on T1-weighted imaging and hyperintense on T2-weighted imaging [70, 71]. Overall, the radiographic features of IPEH in the kidney are nonspecific and often mimic a malignancy with degenerative changes such as necrosis and thrombosis [70].
On gross examination, a well-demarcated hemorrhagic mass with a dark-red to dark-tan cut surface is seen [69, 72]. Microscopically, IPEH is characterized by a well-circumscribed, intravascular proliferation of papillary structures with hyalinized fibrinous cores lined by flat to plump endothelial cells lacking nuclear atypia or mitotic activity [9, 69]. In long-standing lesions, the papillae may fuse to form complex anastomotic-like channels reminiscent of angiosarcoma or anastomosing hemangioma [9]. IPEH may be distinguished from these two entities by its primary intravascular growth and its distinctive morphology of hyalinized fibrinous papillary stalks. In addition, unlike angiosarcoma, IPEH is typically well-circumscribed and shows no pleomorphism, mitotic figures, or areas of necrosis [9].
All reported cases of IPEH in the kidney were treated with partial or radical nephrectomy, due to non-specific clinical and radiologic features leading to pre-operative suspicion for malignancy [69]. The prognosis is excellent, with no reports of malignant transformation or metastasis. One case of local recurrence was reported following partial nephrectomy, which was successfully managed with radical nephrectomy [70].
Hemangioblastoma
Hemangioblastoma is a rare benign tumor of uncertain histogenetic origin which is associated with von Hippel-Lindau (VHL) syndrome, but more often arises sporadically [73]. The vast majority of hemangioblastomas occur in the central nervous system, particularly the cerebellum, but rare cases in extraneural locations have been documented in reports and small series [73]. Hemangioblastoma arising from the kidney is exceedingly rare, with less than 20 cases reported in the literature, and presents a unique and challenging differential diagnosis due to its classic syndromic association with and morphological resemblance to ccRCC [73-79].
Most renal hemangioblastomas arose in older adult patients, with a mean age of 47 years at diagnosis; no sex predilection has been observed. Over half of the reported cases presented as incidental findings; others presented with back or flank pain, hematuria, or polycythemia (a paraneoplastic syndrome associated with cerebellar hemangioblastoma) [15]. The radiologic features of renal hemangioblastoma remain poorly characterized, with CT studies usually displaying a hypodense solid mass, sometimes with a cystic center, which enhances heterogeneously with contrast administration [73, 74]. These findings are typically highly suggestive of RCC, thus prompting nephrectomy for pathologic examination [74].
Grossly, hemangioblastoma presents as a well-circumscribed, red-brown to gray-white solid mass with variable areas of hemorrhage and possible cavitation [15, 73]. Microscopically, hemangioblastoma is composed of sheets of polygonal cells within a complex network of thin-walled blood vessels [73]. The cells contain abundant pale to eosinophilic cytoplasm with fine vacuoles of intracytoplasmic lipid imparting a lipoblast-like appearance, and may focally exhibit a pericytomatous growth pattern around ectatic staghorn-shaped vessels [73, 74]. A few cases have documented the presence of scattered brightly eosinophilic globules both within the cells and in the stroma [73, 74]. The neoplastic cells are generally bland-appearing but may show up to moderate pleomorphism with scattered bizarre cells; however, generally no necrosis and only rare mitotic figures are identified [74]. On immunohistochemical studies, reactivity to alpha-inhibin, neuron-specific enolase (NSE), and S100 is characteristically seen [15]. The vascular network is positive for CD34 and negative for GLUT1 (unlike hemangioblastomas in the central nervous system, which are GLUT1-positive) [73]. Notably, several cases have demonstrated focal reactivity to epithelial antigens (such as EMA and AE1/AE3) and RCC-associated antigens (such as PAX8, CD10, and CAIX), which may confound the differential diagnosis with ccRCC [74, 76, 79].
As previously mentioned, the major differential diagnostic consideration is with ccRCC, as both hemangioblastoma and ccRCC are associated with VHL, show similar morphology with clear cells in a rich vascular background, and may show confounding overlapping immunoreactivity with PAX8, CD10, or CAIX. In fact, reports of “RCC with hemangioblastoma-like features” have also been recently described [80]. General characteristics that can help facilitate the distinction between these two neoplasms include the formation of acinar or alveolar structures in ccRCC, and the positivity for alpha-inhibin, NSE, and S100 in hemangioblastoma [74]. Epithelioid angiomyolipoma also often demonstrates sheets of polygonal cells with prominent vasculature; however, typically the cells show granular cytoplasm (in contrast to the finely vacuolated cytoplasm of hemangioblastoma) and characteristic radial arrangement around thick-walled blood vessels [74]. In addition, the cells are positive for melanocytic markers (HMB-45, Melan-A) and negative for alpha-inhibin. Finally, adrenal cortical carcinoma may also be entertained in the differential as the cells also contain vacuolated cytoplasm and demonstrate positivity for alpha-inhibin; however, this lesion typically shows infiltrative growth with vascular invasion and conspicuous mitotic activity, as well as positivity for adrenal markers such as calretinin and Melan-A [73, 75].
As in other sites, hemangioblastoma in the kidney displays benign behavior, and no reports of recurrence or metastasis have been documented [15]. Nevertheless, patients typically undergo nephrectomy due to pre-operative impression of RCC on imaging studies.
Glomus tumor and variants
The glomus tumor family is a group of pericytic neoplasms derived from the modified perivascular cells of the glomus body, and includes glomus tumor and its variants, glomangioma and glomangiomyoma [81]. Most cases occur in the distal extremities, with rare cases arising in the visceral organs; approximately 30 reports of renal glomus tumor and variants have been documented in the literature to date [81-88]. Patients presented within a wide age range (8 months to 81 years; mean 53 years) with hematuria, abdominal pain, palpable mass, or asymptomatically with an incidentally detected lesion [81]. Interestingly, several cases were associated with hypertension – including in younger patients – which did not improve with resection [81, 86].
The mean size of renal glomus tumors is 4.2 cm, with reported cases measuring from 1.1 to 16 cm. It usually appears as a heterogeneous, hyperechoic mass on sonographic examination, sometimes with peripheral hyperechogenicity and central hypoechogenicity [87-89]. On CT imaging, a well-demarcated, hypodense heterogeneous mass which shows moderate enhancement with contrast administration is typically seen [87, 89]. Overall, no specific radiologic features have been identified and the findings are often suggestive of RCC, necessitating nephrectomy with histopathologic examination for definitive diagnosis [82, 83].
Macroscopically, glomus tumor appears as a well-demarcated, lobular mass with red-brown to gray-white cut surface and variable amounts of hemorrhage [89]. On microscopic exam, typical glomus tumor consists of solid sheets of lobules of monotonous round to ovoid cells with distinct cell borders, within a network of capillary-sized vascular spaces in a myxoid background [81, 89]. Usually no mitotic activity or nuclear atypia is seen, although some cases displayed symplastic changes with scattered bizarre pleomorphic cells (but without increased mitotic activity)[86,89]. Glomangiomas additionally display prominent cavernous blood vessels, and glomangiomyomas feature smooth muscle differentiation with fascicles of spindled myoid cells [81, 84, 89]. Immunohistochemically, the cells of glomus tumor and its variants show positivity for SMA, calponin, caldesmon, and muscle-specific actin; some cases also showed focal positivity for CD34 or desmin [81, 86].
Histopathologic characteristics of malignancy in glomus tumors have not been thoroughly established, and are even less well characterized in renal glomus tumors. Cases designated as malignant glomus tumor, glomus tumor with atypical features, or glomus tumor of uncertain malignant potential in the kidney showed worrisome features such as increased mitotic activity (4-10 mitoses per 50 high-power fields), necrosis, moderate cytologic atypia, focal invasion of the renal capsule, and vascular invasion [81, 84, 87, 88]. Of note, one case of malignant glomus tumor lacked these features and the diagnosis was made based on the presence of metastasis at presentation [90]. In 2001, Folpe et al. defined criteria for malignancy in glomus tumors as visceral location, size greater than 2 cm, atypical mitoses, moderate to high nuclear atypia, and greater than 5 mitoses per 50 high-power fields [91]. However, the most recent World Health Organization classification of soft tissue tumors modified these criteria to include only atypical mitoses and marked nuclear atypia; tumors with visceral location and size greater than 2 cm were downgraded to “uncertain malignant potential.” [40].
The differential diagnosis of glomus tumor in the kidney includes juxtaglomerular cell tumor (JGCT), which may show strikingly similar morphology, but classically presents with hypertension and hypokalemia which resolve with resection [81]. In addition, immunohistochemical studies usually demonstrate positivity for CD34 and CD117, and electron microscopy shows the characteristic rhomboid-shaped renin crystals [81]. Other entities which can be considered in the differential diagnosis include neuroendocrine tumors such as carcinoid and paraganglioma (which can have similar nested architecture and uniform cytology), ccRCC (which also may show epithelioid cells with prominent vasculature), and angiomyolipoma (especially with predominant muscular component) [81, 82]. Immunohistochemical studies with neuroendocrine markers, keratins, and melanocytic markers, respectively, can usually resolve these differential diagnoses.
The prognosis of glomus tumors is typically indolent, but tumors with atypical or malignant features can show more aggressive behavior [81]. Of the six documented cases of renal glomus tumor with atypical or malignant features, two developed metastases: one case with widespread metastases to the lung, liver, skin, brain, and small bowel several years after nephrectomy with death from disease 13 years following diagnosis, and another case with metastasis to the spine present at diagnosis and death from disease 6 months post-operatively [84, 90]. Most renal glomus tumors were managed with complete excision via partial or radical nephrectomy; patients with metastatic disease received limited chemotherapy without apparent improvement in disease [81, 84, 90]. General studies on the treatment of metastatic glomus tumors suggest that chemotherapeutic agents are likely not effective, and the role and future application of systemic therapy in this context remains to be established [92].
Myopericytoma
Myopericytoma is a pericytic tumor originating from perivascular myoid cells, most frequently located in the skin and superficial soft tissue of the extremities [93]. Visceral cases are extremely uncommon; only 12 cases of renal myopericytoma have been reported in the literature to date [94-99]. Myopericytoma tends to occur in middle-aged adults (mean age 49 years). Most patients were asymptomatic, while some patients reported symptoms such as palpable mass and flank pain [95]. These tumors were overall larger than myopericytomas of the skin and soft tissue, with a mean size of 5.6 cm compared to less than 2 cm for more superficial tumors [95].
The imaging characteristics of myopericytoma have not been well established. Ultrasound exam demonstrates a well-circumscribed, hypoechoic, heterogeneous mass with prominent vascularity on color Doppler ultrasonography [95]. CT studies usually show a well-demarcated hypodense solid lesion with heterogeneous contrast enhancement, and sometimes peripheral enhancement with central non-enhancement [95, 97, 99]. The overall findings are similar to those of RCC, leading to frequent misdiagnosis on imaging studies [98, 99].
The gross appearance of myopericytoma is typically of a well-circumscribed but non-encapsulated mass with a tan-red to gray-white cut surface [15, 93]. Microscopically, a mass composed of numerous slit-like thin-walled vascular spaces showing a unique concentric proliferation of plump bland-appearing spindle-shaped cells around the vascular lumina is seen [15, 93]. Many tumors also displayed a wide variety of other morphologic patterns, including areas resembling glomus tumor with nests of uniform round epithelioid cells, angioleiomyoma with fascicles of smooth muscle cells, myofibroma with spindled cells arranged around ectatic thin-walled vessels, fibroma with scattered bland spindled cells within an abundant collagenous stroma, or cavernous hemangioma with prominent dilated vascular spaces [94, 99]. While no nuclear atypia, necrosis, or mitotic activity was seen in most cases, one reported case showed symplastic-type atypia and another case displayed mild atypia with an infiltrative border and was diagnosed as myopericytoma of uncertain malignant potential [93, 94]. On immunohistochemistry, myopericytomas demonstrate positivity for SMA, h-caldesmon, and muscle-specific actin; some cases also show weak activity for desmin or CD34 [15, 94].
The differential diagnosis of myopericytoma includes other pericytic tumors such as glomus tumor, and angioleiomyoma, and myofibroma. As described above, many cases of myopericytoma displayed overlapping histologic features with these other lesions, and in fact many believe that these tumors do not represent distinct entities but rather a morphologic continuum of perivascular myoid neoplasms [94]. Nevertheless, only myopericytoma displays the distinctive concentric perivascular growth pattern of neoplastic cells, a feature that can help distinguish it from other pericytic tumors [94]. Angiomyolipoma is also characterized by abundant blood vessels with perivascular arrangement of cells radiating from the vessel walls which may mimic myopericytoma; however, unlike myopericytoma, angiomyolipoma is typically positive for desmin as well as for melanocytic markers [93, 97]. Finally, benign vascular tumors such as hemangioma or anastomosing hemangioma may exhibit variably abundant stroma containing myoid-like spindled cells which are SMA-positive, but this finding should not be over-interpreted as a feature of myopericytoma [94].
The prognosis of myopericytoma in the kidney appears to be favorable, with no reports of recurrence or metastasis to date [95]. A few cases of malignant myopericytoma in the soft tissue have been documented, but none have been reported in the kidney, to the best of our knowledge. All cases were treated by partial or total nephrectomy, with tumors less than 4 cm generally treated by partial nephrectomy and tumors greater than 4 cm treated with total nephrectomy [94].
Juxtaglomerular cell tumor
Juxtaglomerular cell tumor (JGCT) is a rare renin-secreting neoplasm derived from modified smooth muscle cells of the juxtaglomerular apparatus within the walls of the glomerular arterioles [15]. It is now widely recognized as a rare cause of hypertension and secondary hyperaldosteronism, with approximately 170 cases reported in the literature to date [100]. JGCT typically occurs in young adults; it is twice as common in women and has a peak incidence in the second to third decade of life [15, 101]. Most cases present with hypertension and its related symptoms (such as headache, dizziness, nausea and vomiting) along with hypokalemia, but atypical JGCTs (presenting with hypertension and normokalemia) and very rarely, nonfunctioning JGCTs (presenting with normotension and normokalemia) have also been reported [15, 102, 103]. JGCTs tend to be small tumors, with most under 3 cm; the degree of hypertension is not associated with tumor size [15].
On ultrasonography, JGCT appears as a solid hypoechoic mass [103, 104]. CT studies typically show a well-circumscribed, isoattenuating to hypoattenuating cortical-based tumor without significant enhancement on contrast administration; the findings may raise suspicion for RCC or angiomyolipoma, or in the case of isodense non-enhancing tumors, may go completely undetected on CT imaging [100, 103-105]. MRI of JGCTs usually demonstrates isointensity on T1-weighted images and variable hyperintensity on T2-weighted images with mild heterogeneous contrast enhancement [100, 103]. Overall the imaging features of JGCTs are largely non-specific and cannot be distinguished from other solid renal neoplasms [104].
Macroscopically, JGCT presents as an encapsulated lesion composed of white-gray nodules, occasionally with foci of cystic degeneration [15, 102, 103]. On histopathologic examination, a well-circumscribed mass composed of sheets or trabeculae of uniform round to polygonal cells with a prominent intervening network of thin to thick-walled vascular spaces is seen [15, 103]. Focal areas of hemangiopericytoma-like growth with cells surrounding dilated antler-shaped vessels may be present; areas of cystic, tubular, or papillary architecture may also be observed [15, 103]. Some cases have shown features such as capsular or vascular invasion and focal moderate nuclear atypia; rare cases of metastatic JGCT have also shown variable amounts of necrosis [103, 106].
On immunohistochemistry, JGCTs are consistently positive for CD34, and are variably positive for CD117, SMA, desmin, and caldesmon [15]. JGCTs also show characteristic reactivity with renin, but renin positivity has also been observed in other renal tumors including RCC, Wilm’s tumor, and oncocytoma (which may also lead to hypertension) so care should be taken to not misinterpret this finding as evidence of JGCT [103, 105]. Electron microscopy demonstrates intracytoplasmic rhomboid-shaped crystals corresponding to the renin proto-granules of the neoplastic cells [103]. Molecular genetic studies of JGCT have shown variable chromosomal gains and losses; one study demonstrated monosomy 9 in four of six cases, potentially implicating tumor suppressor genes located on chromosome 9 in the pathogenesis of this tumor [107].
The histopathologic differential diagnosis of JGCT includes glomus tumor, which mimics JGCT closely morphologically but usually contains more abundant myxoid or edematous stroma [103, 107]. In addition, glomus tumor is generally negative for CD34, CD117, and renin on immunohistochemistry. Solitary fibrous tumor (SFT) may also be considered, especially as JGCT often features SFT-like areas of pericytomatous growth; however, SFT does not exhibit the polygonal cells of JGCT and, while CD34-positive like JGCT, SFT has a different overall immunoprofile (CD117 and renin-negative; STAT6, CD99, and BCL2-positive) [103, 107]. Other entities which may enter the differential diagnosis include angiomyolipoma, metanephric adenoma, and papillary RCC (especially for JGCTs showing prominent papillary architecture) [103].
The mainstay of management of JGCT is complete tumor resection, which usually leads to normalization of blood pressure and renin levels [103]. Most JGCTs display benign behavior, but nevertheless patients can experience significant morbidity and complications related to hypertension including retinopathy, congestive heart failure, renal failure, intestinal ischemia, and brain hemorrhage [101, 103, 105, 106]. Three cases of metastatic JGCT have been reported; two cases documented bilateral lung metastasis and one case documented synchronous liver and spleen metastasis at presentation [102, 106, 108]. All three patients were still alive at last reported follow-up (up to 3 years); one patient received several different chemotherapeutic regimens with progression of disease, suggesting that chemotherapy does not appear to provide clinical benefit in the management of these cases [106].
Renal vascular tumors represent a heterogeneous group of neoplasms with a wide array of morphologic characteristics and divergent clinical behavior ranging from benign and indolent to overtly malignant and aggressive. Due to their relative obscurity in comparison to epithelial tumors such as RCC, they are often not considered as diagnostic possibilities and are therefore under-recognized on histopathologic exam. In addition, common renal tumors such as RCC also show a prominent associated vascular proliferation, thus creating increased difficulties in the diagnosis of true vascular-derived neoplasms. Furthermore, on radiography, vascular tumors are often indistinguishable from RCC, leading to potentially unnecessary nephrectomies in the case of benign tumors for histopathologic diagnosis. Future studies of the imaging characteristics of vascular tumors, particularly the specific features of benign vascular tumors, can facilitate an accurate pre-operative diagnosis and spare patients unnecessary nephrectomies. In addition, the management of malignant vascular tumors following nephrectomy is still controversial and not well established in many cases. Therefore, more experience in treating these cases with close, long-term clinical follow-up is needed. Overall, more refined and comprehensive characterization of the distinguishing histopathologic features, imaging findings, and potential therapeutic modalities for renal vascular lesions will lead to future advancement and optimization in the diagnosis and treatment of this group of tumors.
Ethical policy
No research involving experimentation on human or animal subjects was conducted.
Author contributions
MSL performed the literature review and wrote the manuscript. JYR conceived the project and directed the organization and content of the manuscript. All authors read and approved the final manuscript.
Competing interests
All authors declare no competing interests.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Acknowledgements
The authors would like to thank Sasha Pejerrey for her editorial assistance and Subhendu Chakraboty for his assistance with figure preparation.
1. Montgomery E, Epstein JI: Anastomosing hemangioma of the genitourinary tract: a lesion mimicking angiosarcoma. Am J Surg Pathol 2009, 33(9): 1364-1369.
2. Brown JG, Folpe AL, Rao P, Lazar AJ, Paner GP, Gupta R, Parakh R, Cheville JC, Amin MB: Primary vascular tumors and tumor-like lesions of the kidney: a clinicopathologic analysis of 25 cases. Am J Surg Pathol 2010, 34(7): 942-949.
3. Gupta NP, Kumar P, Goel R, Dinda AK: Renal sinus hemangioma simulating renal mass: a diagnostic challenge. Int Urol Nephrol 2004, 36(4): 485-487.
4. Katabathina VS, Vikram R, Nagar AM, Tamboli P, Menias CO, Prasad SR: Mesenchymal neoplasms of the kidney in adults: imaging spectrum with radiologic-pathologic correlation. Radiographics 2010, 30(6): 1525-1540.
5. Bui TL, Glavis-Bloom J, Liu HK, Ushinsky A, Souccar S, Ibe IO, Sasani A, Houshyar R: Multiple renal capillary hemangiomas in a patient with end-stage renal disease. Radiology Case Rep 2019, 14(6): 750-754.
6. Leak BJ, Javidan J, Dagher R: A rare case of renal hemangioma presenting as polycythemia. Urology 2001, 57(5): 975.
7. Memmedoğlu A, Musayev J: Spontaneous rupture of the kidney in the patients with synchronous renal hemangioma and nephrogenic hypertension. Turk J Urol 2015, 41(4): 231.
8. Beamer M, Love M, Ghasemian S: Renal capillary haemangioma associated with renal cell carcinoma and polycythaemia in acquired cystic disease. BMJ Case Rep 2017, 2017.
9. Mehta V, Ananthanarayanan V, Antic T, Krausz T, Milner J, Venkataraman G, Picken MM: Primary benign vascular tumors and tumorlike lesions of the kidney: a clinicopathologic analysis of 15 cases. Virchows Arch 2012, 461(6): 669-676.
10. Fellegara G, Rosai J: Multifocal capillary hemangioma-like vascular proliferation of the kidney associated with clear cell renal cell carcinoma: a case report and review of the literature. Int J Surg Pathol 2013, 21(4): 424-426.
11. Vasquez E, Aulivola B, Picken M, Milner J: Capillary hemangioma masquerading as a renal artery pseudoaneurysm. Urology 2012, 79(5): 987-989.
12. Geramizadeh B, Shams N, Iranpour P, Rajabi MJ: Renal Capillary Hemangioma Mimicking Urothelial Carcinoma, A Case Report and Review of the Literature. Iran J Pathol 2019, 14(2): 175.
13. Campistol JM, Agusti C, Torras A, Campo E, Abad C, Revert L: Renal hemangioma and renal artery aneurysm in the Klippel-Trenaunay syndrome. J Urology 1988, 140(1): 134-136.
14. Schofield D, Zaatari GS, Gay BB: Klippel-Trenaunay and Sturge-Weber syndromes with renal hemangioma and double inferior vena cava. J Urology 1986, 136(2): 442-445.
15. Samaratunga H, Delahunt B: Mesenchymal tumors of adult kidney. Semin Diagn Pathol 2015, 32(2): 160-171.
16. Schlomer BJ: Gross hematuria in infancy: a case of renal hemangioma managed endoscopically. Urology 2017; 101: 166-168.
17. Raman SP, Hruban RH, Fishman EK: Beyond renal cell carcinoma: rare and unusual renal masses. Abdom Radiol 2012, 37(5): 873-874.
18. Lee HS, Koh BH, Kim JW, Kim YS, Rhim HC, Cho OK, Hahm CK, Woo YN, Park MH: Radiologic findings of renal hemangioma: report of three cases. Korean J Radiol 2000, 1(1): 60-63.
19. Le O, Roy A, Silverman PM, Kundra V: Common and uncommon adult unilateral renal masses other than renal cell carcinoma. Cancer Imaging 2012, 12(1): 194.
20. Prasad SR, Humphrey PA, Menias CO, Middleton WD, Siegel MJ, Bae KT, Heiken JP: Neoplasms of the renal medulla: radiologic-pathologic correlation. Radiographics 2005, 25(2): 369-380.
21. Cheon PM, Rebello R, Naqvi A, Popovic S, Bonert M, Kapoor A: Anastomosing hemangioma of the kidney: radiologic and pathologic distinctions of a kidney cancer mimic. Curr Oncol 2018, 25(3): 220-223.
22. Chen T, Jin P, Wang Y, Zhao X, Yang L: Renal cavernous hemangioma in a child: case report and review of the literature. Urology 2011, 78(5): 1185-1186.
23. Taneja K, Arora S, Rogers CG, Gupta NS, Cheng L, Williamson SR: Unclassified hemangioma-like renal cell carcinoma: a potential diagnostic pitfall. Hum Pathol 2018, 75: 132-136.
24. Kim EK, Jang M, Choi YJ, Cho NH: Renal Cell Carcinoma with Hemangioma-Like Features: Diagnostic Implications and Review of the Literature. Int J Surg Pathol 2019, 27(6): 631-638.
25. Lapham RL, Ro JY, Ordonez NG, Ayala AG: Angiomatoid renal cell carcinoma: report of four cases of a previously undescribed histological pattern. Lab Invest 1997, 76(1): 454.
26. Daneshmand S, Huffman JL: Endoscopic management of renal hemangioma. J Urology 2002, 167(2): 488-489.
27. Sethi S, Agarwal V, Chopra P: Cavernous hemangioma of the kidney: A report of two cases and review of the literature. Urol Ann 2012, 4(3): 187.
28. Göğüş Ç, Kılıç S, Ataoğlu Ö, Göğüş O: Large cavernous hemangioma of the kidney presenting as a solid renal mass. Int Urol Nephrol 2001, 33(4): 615-616.
29. Omiyale AO: Anastomosing hemangioma of the kidney: a literature review of a rare morphological variant of hemangioma. Ann Transl Med 2015, 3(11): 151.
30. Lappa E, Drakos E: Anastomosing Hemangioma: Short Review of a Benign Mimicker of Angiosarcoma. Arch Pathol Lab Med 2019.
31. Lin M, Ngo T, Schwartz MR, Mehta RR, Ayala AG, Ro JY: Anastomosing hemangioma of breast: an unusual case at an unusual site. J Breast Cancer 2020. In press.
32. Perdiki M, Datseri G, Liapis G, Chondros N, Anastasiou I, Tzardi M, Delladetsima JK, Drakos E: Anastomosing hemangioma: report of two renal cases and analysis of the literature. Diagn Pathol 2017, 12(1): 14.
33. Kryvenko ON, Haley SL, Smith SC, Shen SS, Paluru S, Gupta NS, Jorda M, Epstein JI, Amin MB, Truong LD: Haemangiomas in kidneys with end‐stage renal disease: a novel clinicopathological association. Histopathology 2014, 65(3): 309-318.
34. Büttner M, Kufer V, Brunner K, Hartmann A, Amann K, Agaimy A: Benign mesenchymal tumours and tumour-like lesions in end-stage renal disease. Histopathology 2013, 62(2): 229-236.
35. Berker NK, Bayram A, Tas S, Bakir B, Caliskan Y, Ozcan F, Kilicaslan I, Ozluk Y: Comparison of Renal Anastomosing Hemangiomas in End-Stage and Non–End-Stage Kidneys: A Meta-Analysis with a Report of 2 Cases. Int J Surg Pathol 2017, 25(6): 488-496.
36. Al-Maghrabi HA, Al Rashed AS: Challenging pitfalls and mimickers in diagnosing anastomosing capillary hemangioma of the kidney: case report and literature review. Am J Case Rep 2017, 18: 255-262.
37. O’Neill AC, Craig JW, Silverman SG, Alencar RO: Anastomosing hemangiomas: locations of occurrence, imaging features, and diagnosis with percutaneous biopsy. Abdom Radiol 2016, 41(7): 1325-1332.
38. Rodrigues MA, Fonseca EK, Yamauchi FI, Baroni RH: Anastomosing hemangioma simulating renal cell carcinoma. Int Braz J Urol 2017, 43(5): 987-989.
39. Bean GR, Joseph NM, Gill RM, Folpe AL, Horvai AE, Umetsu SE: Recurrent GNAQ mutations in anastomosing hemangiomas. Modern Pathol 2017, 30(5): 722-727.
40. Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F, editors: World Health Organization classification of tumours of soft tissue and bone, 4th edition. Lyon: IARC Press, 2013.
41. Karasavvidou F, Barbanis S, Gravas S, Ioannou M, Oeconomou A, Pappa D, Melekos MD, Koukoulis G: Primary renal epithelioid hemangioendothelioma. Oncol Res Treat 2009, 32(4): 203-205.
42. Shin DH, Chen M, Niemeier LA: Primary epithelioid hemangioendothelioma of the kidney and penis. Can J Urol 2010, 17(6): 5480-5482.
43. Tolkach Y, Petrov S, Lerut E, Van Poppel H: Epithelioid hemangioendothelioma of the kidney treated with sunitinib. Oncol Res Treat 2012, 35(6): 376-378.
44. Xiao Y, Wang C, Song Y, Wang C, Wang X, Zhang X, Xu A: Primary epithelioid hemangioendothelioma of the kidney: the first case report in a child and literature review. Urology 2013, 82(4): 925-927.
45. Liu Y, Liu A, Wu J, Liu T: Epithelioid hemangioendothelioma arising from the kidney: A rare case report. Medicine 2019, 98(34).
46. Roy S, Parwani AV: Primary renal epithelioid hemangioendothelioma. Case Rep Pathol 2012; 2012.
47. Indolfi P, Donofrio V, Fusco C, Di Martino M, Di Pinto D, Indolfi C, Scanderbech AM, Casale F: Kaposiform hemangioendothelioma of the kidney: an unusual presentation of a rare vascular neoplasm. J Pediatr Hematol Onc 2010; 32(5): 195-198.
48. Zhang J, Wu B, Zhou GQ, Zhang RS, Wei X, Yu B, Lu ZF, Ma HH, Shi QL, Zhou XJ: Composite hemangioendothelioma arising from the kidney: case report with review of the literature. Int J Clin Exp Pathol 2013, 6(9): 1935.
49. Omiyale AO, Carton J: Clinical and pathologic features of primary angiosarcoma of the kidney. Curr Urol Rep 2018, 19(2): 4.
50. Li XL, Tian X, Guo J, Zhang H, Wang S, Dai SD: Primary renal angiosarcoma: A case report and literature review. Prec Radiat Oncol 2018, 2(3): 94-101.
51. Omiyale AO: Clinicopathological features of primary angiosarcoma of the kidney: a review of 62 cases. Transl Androl Urol 2015, 4(4): 464-73.
52. Darlington D, Anitha FS: Primary Renal Angiosarcoma Mimicking Renal Cell Carcinoma: A Case Report. Cureus 2019, 11(1).
53. Boni A, Cochetti G, Sidoni A, Bellezza G, Lepri E, De Giglio A, Turco M, De Vermandois JA, Del Zingaro M, Cirocchi R, Mearini E: Primary angiosarcoma of the kidney: case report and comprehensive literature review. Open Med 2019, 14(1): 443-455.
54. Heo SH, Shin SS, Kang TW, Kim GE: Primary renal angiosarcoma with extensive hemorrhage: CT and MRI findings. Int Braz J Urol 2019, 45(2): 402-405.
55. Waqas M, Rahim W, Shohab D, Khawaja MA, Ali Z, Mamoon N: Primary Renal Epithelioid Angiosarcoma. JCPSP-J Coll Physici 2018, 28(3): 66-68.
56. Samadi K, Arellano RS: Primary perirenal angiosarcoma: A rare presentation of a perirenal mass. Radiol Case Rep 2019, 14(1): 28-29.
57. Lodhi HT, Inayat F, Munir A, Ilyas G: Primary renal angiosarcoma: a diagnostic and therapeutic challenge. BMJ Case Rep 2018.
58. Celebi F, Pilanci KN, Saglam S, Balci NC: Primary renal angiosarcoma with progressive clinical course despite surgical and adjuvant treatment: A case report. Oncol Lett 2015, 9(4): 1937-1939.
59. Detorakis EE, Chryssou E, Raissaki M, Androulidakis E, Heretis I, Haniotis V, Karantanas A: Primary renal angiosarcoma: radiologic-pathologic correlation and literature review. Tumori J 2013, 99(3): 111-116.
60. Mukendi AM, Rauf A, Doherty S, Mahlobo F, Afolayan P, Dawadi S: Renal arteriovenous malformation: An unusual pathology. SA J Radiol 2019, 23(1): 5.
61. Cura M, Elmerhi F, Suri R, Bugnone A, Dalsaso T: Vascular malformations and arteriovenous fistulas of the kidney. Acta Radiol 2010, 51(2): 144-149.
62. Hatzidakis A, Rossi M, Mamoulakis C, Kehagias E, Orgera G, Krokidis M, Karantanas A: Management of renal arteriovenous malformations: a pictorial review. Insights Imaging 2014, 5(4): 523-530.
63. Eom HJ, Shin JH, Cho YJ, Nam DH, Ko GY, Yoon HK: Transarterial embolisation of renal arteriovenous malformation: safety and efficacy in 24 patients with follow-up. Clin Radiol 2015, 70(11): 1177-1184.
64. Choi YA, Park BK, Kim CK, Park SY: MRI features of a solid mass-like renal lymphangioma: case report. Clin Imaging 2012, 36(4): 398-401.
65. Chaabouni A, Rebai N, Fourati M, Rekik S, Chabchoub K, Slimen MH, Bahloul A, Mhiri MN: Cystic lymphangioma of the kidney: diagnosis and management. Int J Surg Case Rep 2012, 3(12): 587-589.
66. Honma I, Takagi Y, Shigyo M, Sunaoshi K, Wakabayashi J, Harada O, Miyao N: Lymphangioma of the kidney. Int J Urol 2002, 9(3): 178-182.
67. Lal H, Agarwal V, Naik S: Renal lymphangioma. J Integr Nephrol Androl 2016, 3(4): 130-132.
68. Chua N, Wolfe K, Mehta S, Lodge RN, Liyanage SH: Triparametric ultrasound in differentiating multicystic renal masses: a rare presentation of unilateral focal renal lymphangioma. Radiol Case Rep 2017, 12(4): 731-737.
69. Essid MA, Bouzouita A, Blel A, Gharbi M, Chakroun M, Miled AB, Ayed H, Cherif M, Slama MR, Derouiche A, Chebil M: Masson’s tumor of the kidney: a case report. J Med Case Rep 2018, 12(1): 376.
70. Alkan E, Sağlıcan Y, Özkanlı AO, Balbay MD: The first recurrent intravascular papillary endothelial hyperplasia (Masson’s tumor) of the kidney. Turk J Urol 2016, 42(3): 202-205.
71. Moreillo-Vicente L, Gimenez-Bachs JM, Agusti-Martinez A, Salinas-Sanchez AS: Intravascular Papillary Endothelial Hyperplasia: Regarding a Case. Ann Clin Case Rep 2017, 2: 1319.
72. Van den Bogaert S, Boel K, Van Poppel H, Oyen R, Van Damme B: Masson’s tumour of the kidney. Cancer Imaging 2002, 2(2): 116-119.
73. Ip YT, Yuan JQ, Cheung H, Chan JK: Sporadic hemangioblastoma of the kidney: an underrecognized pseudomalignant tumor? Am J Surg Pathol 2010, 34(11): 1695-1700.
74. Zhao M, Williamson SR, Yu J, Xia W, Li C, Zheng J, Zhu Y, Sun K, Wang Z, Cheng L: PAX8 expression in sporadic hemangioblastoma of the kidney supports a primary renal cell lineage: implications for differential diagnosis. Hum Pathol 2013, 44(10): 2247-2255.
75. Wang Y, Wei C, Mou L, Zhang Q, Cui Z, Li X, Ye J, Lai Y: Sporadic renal haemangioblastoma: Case report and review of the literature. Oncol Lett 2013, 5(1): 360-362.
76. Jiang JG, Rao Q, Xia QY, Tu P, Lu ZF, Shen Q, Zhang RS, Yu B, Zhou XJ, Shi SS, Shi QL: Sporadic hemangioblastoma of the kidney with PAX2 and focal CD10 expression: report of a case. Int J Clin Exp Pathol 2013, 6(9): 1953.
77. Doyle LA, Fletcher CD: Peripheral hemangioblastoma: clinicopathologic characterization in a series of 22 cases. Am J Surg Pathol 2014, 38(1): 119-127.
78. Wu Y, Wang T, Zhang PP, Yang X, Wang J, Wang CF: Extraneural hemangioblastoma of the kidney: the challenge for clinicopathological diagnosis. J Clin Pathol 2015, 68(12): 1020-1025.
79. Kuroda N, Agatsuma Y, Tamura M, Martinek P, Hes O, Michal M: Sporadic renal hemangioblastoma with CA9, PAX2 and PAX8 expression: diagnostic pitfall in the differential diagnosis from clear cell renal cell carcinoma. Int J Clin Exp Pathol 2015; 8(2): 2131.
80. Montironi R, Lopez-Beltran A, Cheng L, Galosi AB, Montorsi F, Scarpelli M: Clear cell renal cell carcinoma (ccRCC) with hemangioblastoma-like features: a previously unreported pattern of ccRCC with possible clinical significance. Eur Urol 2014, 66(5): 806-810.
81. Sirohi D, Smith SC, Epstein JI, Balzer BL, Simko JP, Balitzer D, Benhamida J, Kryvenko ON, Gupta NS, Paluru S, da Cunha IW: Pericytic tumors of the kidney—a clinicopathologic analysis of 17 cases. Hum Pathol 2017; 64: 106-117.
82. Almaghrabi A, Almaghrabi N, Al-Maghrabi H: Glomangioma of the kidney: a rare case of glomus tumor and review of the literature. Case Rep Pathol 2017, 1-7.
83. Dee E, Loghin A, Toth T, N?znean A, Borda A: Glomus Tumor of the Kidney: Case report. Acta Medica Marisiensis 2018, 64(3): 126-129.
84. Li R, Petros FG, Davis CJ, Ward JF: Characterization of Glomus Tumors of the Kidney. Clin Genitourin Cancer 2018, 16(1): 253-256.
85. Novis E, Raman A, Maclean F, Lazzaro E: Glomus tumour of the kidney: a case report and review of the literature. ANZ J Surg 2018, 88(6): 653-654.
86. Zhao M, Wang AX, Zhu X, Yu JJ, Wang W, Zhang DH, He XL, He HY, Teng XD: Clinicopathologic features of glomus tumor of the kidney. Chinese J Pathol 2018, 47(8): 580-584.
87. Zhao M, Yang M, Gu W, Chen X, Chen H, Kuick CH, Chang KT, Tang H: Glomus Tumor of the Kidney in a Child with Tuberous Sclerosis. Pediatr Dev Pathol 2019, 1-5.
88. Chen YA, Li HN, Wang RC, Hung SW, Chiu KY: Malignant glomus tumor of the kidney: a case report and review of the literature. Clin Genitourin Cancer 2017, 15(1): 151-153.
89. Al-Ahmadie HA, Yilmaz A, Olgac S, Reuter VE: Glomus tumor of the kidney: a report of 3 cases involving renal parenchyma and review of the literature. Am J Surg Pathol 2007, 31(4): 585-591.
90. Lamba G, Rafiyath SM, Kaur H, Khan S, Singh P, Hamilton AM, Ang DC: Malignant glomus tumor of kidney: the first reported case and review of literature. Hum Pathol 2011, 42(8): 1200-1203.
91. Folpe AL, Fanburg–Smith JC, Miettinen M, Weiss SW: Atypical and malignant glomus tumors: analysis of 52 cases, with a proposal for the reclassification of glomus tumors. Am J Surg Pathol 2001, 25(1): 1-12.
92. Jin WS, Jeon IS: A Case of Malignant Glomus Tumor on Shoulder with Pulmonary Metastasis Treated with Doxorubicin and Ifosfamide in a 10-year Old Girl. Clin Pediatr Hematol Oncol 2016, 23(1): 65.
93. Lau SK, Klein R, Jiang Z, Weiss LM, Chu PG: Myopericytoma of the kidney. Hum Pathol 2010, 41(10): 1500-1504.
94. Li J, Zhao M, Chen Z, Zou L, Teng X: Renal myopericytoma: a clinicopathologic study of six cases and review of the literature. Int J Clin Exp Pathol 2015; 8(5): 4307.
95. Qiao Y, Zhou Z, Yan W, Xiao Y, Zhao B, Ji Z, Li H: Renal myopericytoma: a case report and literature review. Int J Clin Exp Med 2018; 11(5): 5193-5199.
96. Yang SC, Wang Z, Lin ZW, Huang LL, Luo CJ, Wang LT: Case Report Renal myopericytoma with nephrotic syndrome: report of a case and review of literature. Int J Clin Exp Pathol 2016; 9(2): 2443-2450.
97. Zhang Z, Yu D, Shi H, Xie D: Renal myopericytoma: A case report with a literature review. Oncol Lett 2014, 7(1): 285-287.
98. Dhingra S, Ayala A, Chai H, Moreno V, Zhao B: Renal myopericytoma: case report and review of literature. Arch Pathol Lab Med 2012, 136(5): 563-566.
99. Zhao M, Williamson SR, Sun K, Zhu Y, Li C, Xia W, Qi H, Wang L, Linos K, Cheng L: Benign perivascular myoid cell tumor (myopericytoma) of the urinary tract: a report of 2 cases with an emphasis on differential diagnosis. Hum Pathol 2014, 45(5): 1115-1121.
100. Faucon AL, Bourillon C, Grataloup C, Baron S, Bernadet-Monrozies P, Vidal-Petiot E, Azizi M, Amar L: Usefulness of magnetic resonance imaging in the diagnosis of juxtaglomerular cell tumors: a report of 10 cases and review of the literature. Am J Kidney Dis 2019, 73(4): 566-571.
101. Inam R, Gandhi J, Joshi G, Smith NL, Khan SA: Juxtaglomerular Cell Tumor: Reviewing a Cryptic Cause of Surgically Correctable Hypertension. Curr Urol 2019, 13(1): 7-12.
102. Huang PW, Lin YC, Wu KF, Sheng TW, Su PJ: Juxtaglomerular cell tumor with lung metastases in a young male patient. J Cancer Res Pract 2019, 6(3): 128.
103. Kuroda N, Gotoda H, Ohe C, Mikami S, Inoue K, Nagashima Y, Petersson F, Alvarado-Cabrero I, Pan CC, Hes O, Michal M: Review of juxtaglomerular cell tumor with focus on pathobiological aspect. Diagn Pathol 2011, 6(1): 80.
104. Yang H, Wang Z, Ji J: Juxtaglomerular cell tumor: A case report. Oncol Lett 2016, 11(2): 1418-1420.
105. Kim HJ, Kim CH, Choi YJ, Ayala AG, Amrikachi M, Ro JY: Juxtaglomerular cell tumor of kidney with CD34 and CD117 immunoreactivity: report of 5 cases. Arch Pathol Lab Med 2006, 130(5): 707-711.
106. Cucchiari D, Bertuzzi A, Colombo P, De Sanctis R, Faucher E, Fusco N, Pellegrinelli A, Arosio P, Angelini C: Juxtaglomerular cell tumor: multicentric synchronous disease associated with paraneoplastic syndrome. J Clin Oncol 2013, 31(14): 240-242.
107. Kuroda N, Maris S, Monzon FA, Tan PH, Thomas A, Petersson FB, Gatalica Z, Ghazalpour A, Bender RP, Grossmann P, Michal M: Juxtaglomerular cell tumor: a morphological, immunohistochemical and genetic study of six cases. Human Pathol 2013, 44(1): 47-54.
108. Duan X, Bruneval P, Hammadeh R, Fresco R, Eble JN, Clark JI, Vigneswaran WT, Flanigan RC, Picken MM: Metastatic juxtaglomerular cell tumor in a 52-year-old man. Am J Surg Pathol 2004, 28(8): 1098-1102.
Annals of urologic oncology
p-ISSN: 2617-7765, e-ISSN: 2617-7773
 Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.