
 Submit Manuscript
Submit Manuscript
Review Article | Open Access
Renal Cell Carcinoma

1Faculty of Medicine, Wuhan University, Wuhan City, Hubei Province, 430071, P.R. China.
2Department of Pathology and Genomic Medicine, Houston Methodist Hospital, Weill Medical College of Cornell University, Houston, TX 77030, U.S.A.
Correspondence: Peng Zhang (Faculty of Medicine, Wuhan University, Donghu Load 115, Wuchang District, Wuhan City, Hubei Province, P.R. China; Email: misiss5@whu.edu.com).
Annals of Urologic Oncology 2018, Volume 1, Issue 1, Pages: 1-18, https://doi.org/10.32948/auo.2018.11.1
Received: 17 Oct 2018 | Accepted: 30 Oct 2018 | Published online: 15 Nov 2018 
The global incidence of cases of kidney cancer has increased rapidly, and a relatively high incidence of kidney cancer has been reported in developed countries such as Northern and Eastern Europe. Various factors can affect the incidence and mortality of kidney cancer, including demographic risk factors, lifestyle factors, iatrogenic risk factors, nutritional factors and diet, occupation, and genetic factors. Renal cell carcinoma (RCC) refers to a tumor group with heterogeneity derived from renal tubular cells, which form almost all kidney cancer types. Clear cell RCC (ccRCC) is the most frequent renal tumor subtype, accounting for 75% of renal cancer, followed by papillar RCC (pRCC) making up approximately 10% of RCC. Hematoxylin-eosin staining shows a clear, eosinophilic cytoplasm in ccRCC cells. Epithelial cells forming the papillae and tubules have pRCC histological characteristics. Traditionally, genetic mutations of VHL and MET are the genetic features in ccRCC and pRCC, respectively. Recently, a new concept supports the contribution of mutations in some chromatin-modifier genes, including polybromo 1 (PBRM1), SET domain containing 2 (SETD2), BRCA1-associated protein-1 (BAP1), and lysine (K)-specific demethylase 5C (KDM5C). The metabolic disease concept in renal cancer is noted by researchers worldwide. The PD-1 pathway has been valued by researchers of kidney cancer in recent years, and new agents, such as anti-PD-1 monoclonal antibodies (nivolumab and pembrolizumab) and CTLA4 inhibitors (Ipilimumab), have been approved to treat advanced RCC. Partial nephrectomy (PN) and radical nephrectomy (RN) remain the standard management option for local RCC with a stage of T1 and T2, respectively. PN can also be selected for T2 stage RCC in suitable cases. Even though targeted therapy consisting of mainly the anti-VEGF and anti-mTOR pathways is recommended as the first-line and second-line treatment for RCC, the effectiveness and side effect of these therapies should be improved in future research.
Key words renal cell carcinoma, review, genetic mutation, management, immune checkpoint
The global incidence of cases of kidney cancer were estimated to be 142,463 in 1990, with the number rapidly rising to 273,518 in 2008 and 294,501 in 2013 [9, 10]. There was a mean global age-standardized incidence rate of 7.75 per 100,000 people per year in 1990. The number remained roughly stable with a mean of 4 in 2008 and 6.71 in 2013 [9, 10]. The increase in the global incidence of cases of kidney cancer could be affected by population growth (35.0%) and the change in age structure (34.7%). Based on data collected from all confirmed cases of kidney cancer diagnosed from 2001 to 2010 in the U.S., the incidence of RCC in males is almost 2 times higher than in females [11]. Global age-standardized incidence rates per 100 000 kidney cancer case in males (mean 6.73) were also almost 2 times greater than in females (mean 2.97) in 2013 [10]. There was positive correlation between age and incidence of kidney cancer. The elderly (age > 75 yr) has the highest incidence of kidney cancer [12]. Black and white individuals have a relatively higher incidence rate of kidney cancer than yellow individuals in the USA [11]. The distribution of the incidence of kidney cancer shows significant differences worldwide. There is a relatively high incidence of kidney cancer distributed in Northern and Eastern Europe, North America, and Australia, and a relatively low incidence is estimated in much of Africa and South-East Asia [10, 13, 14]. Interestingly, black people in Africa have a lower incidence of kidney cancer than black people in the USA. The incidence rate of kidney cancer in developed countries is much higher than in developing countries. The age-standardized incidence rates per 100,000 cases of kidney cancer in both females and males in developing countries is 1.34, and the number rises to 3.27 in developed countries. An increase of 35.8% in the incidence of kidney cancer was reported in both males and females in developed countries from 1990 to 2013. Similarly, an increase of 34.32% was reported in developing countries [10].
There was no obvious change in mortality due to kidney cancer from 1990 to 2013. Global deaths caused by kidney cancer were 77,900, and the age-standardized death rate of kidney cancer was 2.1 in 1990. The global death rate caused by kidney cancer was 134,000 and the age-standardized death rate 2.2 in 2013 [15]. There was a significant difference in kidney cancer mortality rates between developed and developing countries. The age-standardized death rate in developed countries was 3.7 while it was 1.3 in developing countries in 2013 [16]. Additionally, both numbers were 2.8 vs. 1.3 in 2012 [1]. In 2012, Lithuania, the Czech Republic, Latvia and Estonia were reported to have the highest mortality rates, and Micronesia/Polynesia, Middle Africa, Western Africa and South-Central Asia had the lowest death rates due to kidney cancer [1]. Similar to the incidence of kidney cancer, the mortality rate was also affected by sex. The male mortality rate reached up to threefold greater than that of the female rate according to an analysis of global data (2003-2007) [12].
Various risk factors can affect the incidence and mortality of kidney cancer, such as demographic risk factors (race, age, and sex), lifestyle factors (smoking and obesity), iatrogenic risk factors (hypertension and use of antihypertensive medications, acquired renal cystic disease, diabetes and urinary tract infection), nutritional factors and diet, occupation, and genetic factors (Figure 1) [17, 18]. As described above, the distributional difference in the incidence and mortality of kidney cancer directly reflects that race, age and sex have an influence on kidney cancer prevalence. Tobacco continues to be the dominant risk factor in global male kidney cancer deaths, while it is weakly associated with kidney cancer in females. Instead of smoking, obesity is a stronger risk factor for kidney cancer in females than males. A meta-analysis by Callahan et al. showed that obesity (BMI ≥ 30 kg/m2) was associated with ccRCC and chRCC but not with pRCC [19]. Numerous studies have suggested that blood pressure is associated with kidney cancer risk [20, 21]. Recently, a meta-analysis by Khemayanto et al. reported a positive association between hypertension and kidney cancer, and a dose-response analysis revealed that each 10 mmHg increase in systolic blood pressure and diastolic blood pressure were significantly associated with a 10% and 22% increased risk of kidney cancer, respectively [22]. Acquired renal cystic disease is a definite risk factor for RCC, and the period of dialysis may be related to the higher incidence of RCC [23, 24]. Other diseases, including diabetes and urinary tract infection, have also been reported as potential risk factors for RCC, but the conclusions have been controversial [25-27]. A large number of studies involved total fat or various types of fat intake and RCC risk, and their results were inconsistent [28]. Meta-analyses showed that alcohol and fruit and vegetable intake can reduce the risk of kidney cancer [29, 30]. Although RCC is not a typical occupational disease, exposure to some organic compounds, such as trichloroethylene (TCE), can be linked to kidney cancer [31]. A review did not support that occupational exposure to TCE is a risk factor for cancer of any specific site [32]. A corresponding familial syndrome in each of the common histologic subtypes of RCC was caused by a distinct genetic alteration, even though these syndromes are relatively rare [33]. Various gene mutations are associated with RCC, and two typical genetic variations in VHL and MET are involved in occurrence of ccRCC and pRCC, respectively [34, 35]. The specific content of the kidney oncology gene and histologic subtypes will be discussed in subsequent sections of the article.
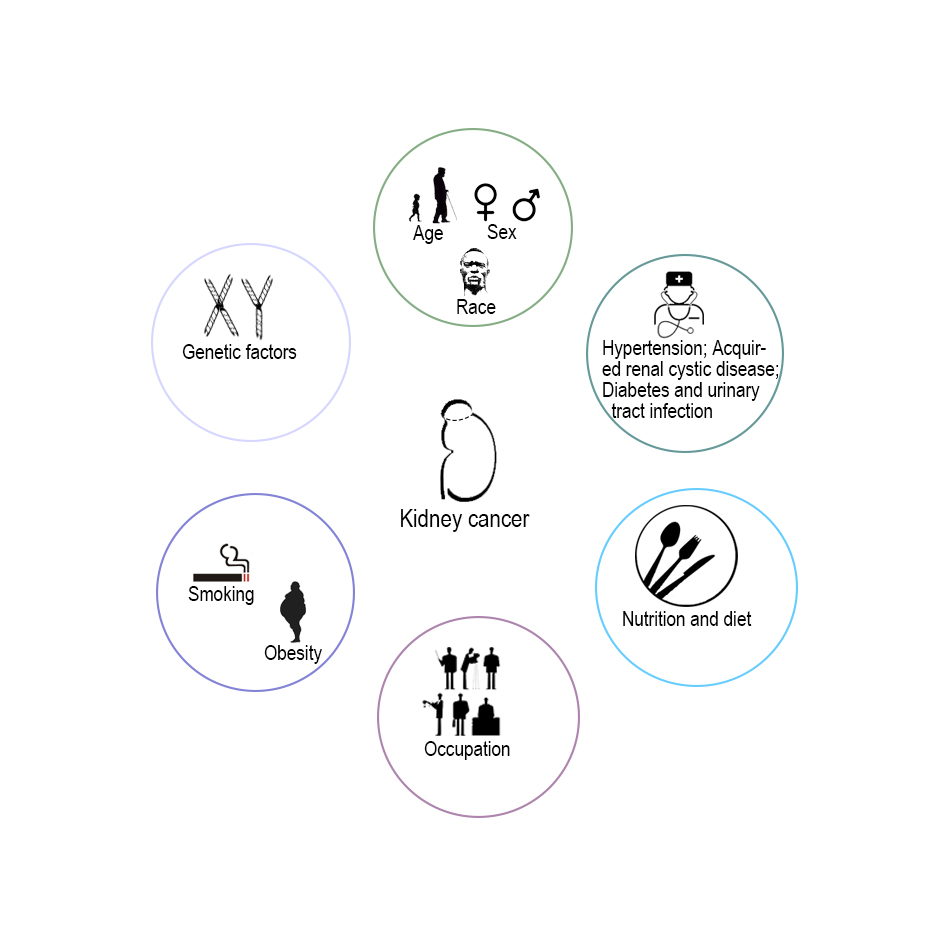 Figure 1. Kidney cancer risk factors.
Figure 1. Kidney cancer risk factors.
RCC refers to a heterogenous tumor group derived from renal tubular cells [36]. According to the morphological and genetic characteristics of RCC described by WHO, there are more than a dozen RCC subtypes [37, 38]. ccRCC, pRCC and chRCC are the three most common types of RCC and consist of approximately 90% RCCs. According to the 2016 WHO classification of kidney tumors, 16 renal cell tumors were classified and five renal tumor entities were newly identified, including hereditary leiomyomatosis and RCC syndrome-associated RCC, succinate dehydrogenase-deficient RCC, tubulocystic RCC, acquired cystic disease-associated RCC and clear cell papillary RCC [38]. Most subtypes of RCC classified by WHO in 2016 are presented in Table 1 [38-40]. Here we emphatically introduce several common types of RCC classified by WHO.
Clear cell renal cell carcinoma (ccRCC)
Clear cell RCC generally consists of solitary cortical neoplasms it occurs equally in either kidney. It is the most frequent renal tumor subtype, making up to 75% of RCCs, and it exhibits a typical golden yellow tumor surface due to the abundant lipid content of the cells (Figure 2A,F). Hematoxylin-eosin staining shows solid alveolar or acinar pattern of tumor cells with a regular network of thin walled sinusoidal blood vessels (Figure 2A).The tumor cells have a round nucleus with evenly distributed chromatin and variablesized nucleolus depending on the tumor grade, and havea clear oreosinophilic cytoplasm [2]. Von Hippel-Lindau (VHL) disease will result in this tumor type, which is a hereditary RCC. The alterations in chromosome regions (-3p,+5q22, -6q, -8p, -9p, -14) are identified as its genetic features [37]. PAX-8 with nuclear staining and CA9 with membranous staining are regarded as useful markers based on immunohistochemical methods [41].
Papillar renal cell carcinoma (pRCC)
pRCC makes up approximately 10% of RCCs, second to ccRCC (Figure 2F) [42-44]. Bilateral and multifocal tumors occur more frequently in pRCC than in other renal malignancies. The tumor tissue, which is usually friable, is bounded by a thick pseudocapsule and frequently shows fibrosis and hemorrhage. Epithelial cells forming the papillae and tubules are its histological characteristics. Two distinct groups, type 1 and type 2, are defined in pRCC. In type 1, papillae are covered by a single raw of low nuclear grade tumor cells (nuclear grade 1 or 2) with scanty cytoplasm (Figure 2B). Type 2 shows a higher nuclear grade (nuclear grade 3 or 4) with abundant eosinophilic cytoplasm and stratified or pseudostratified nuclei (Figure 2C) [2]. Hereditary papillary RCC will result in type 1 pRCC with mutation of the c-MET gene and hereditary leiomyomatosis, and renal cell cancer syndromes (HLRCC) with mutation of the fumarate hydratase gene are associated with type 2 pRCC. The alteration in chromosome regions (+3q,+7,+8,+12,+16,+17,+20, -Y) has been identified as the genetic features [37].
Chromophobe renal cell carcinoma (chRCC)
ChRCC accounts for approximately 5% of RCC (Figure 2F) [45]. Solitary tumors commonly occur with a homogeneous gray or gray-brown gross appearance. Compared with ccRCC and pRCC, this type of RCC has a better prognosis and a mortality less than 10% [45]. These tumors consist of large polygonal cells with a reticulated cytoplasm and prominent cell membranes, reisinoid wrinkled nuclei and perinuclear claring (Figure 2D). They must be differentiated from those of oncocytoma due to the eosinophilic variation among the two types of renal cancer. Birt-Hogg-Dubé (BHD) syndrome is related to chRCC with mutation of the BHD gene [2]. The alteration of chromosome regions (-1, -2, -6, -10, -17, -21, hypodiploidy) has been identified as its genetic feature [37].
Collecting duct carcinoma
This is an extremely aggressive renal tumor type accounting for < 1% of all renal cancer types. It usually occurs in the central region of the kidney and exhibits a firm, gray-white appearance with irregular borders. Histologically, it has a tubulo-papillary architecture with a high nuclear grade and a characteristic desmoplastic stromal reaction (Figure 2E) [2]. The alteration of chromosome regions (-1q, -6p, -8p, -13q, -21q, -3p) has been identified as its genetic feature [37].
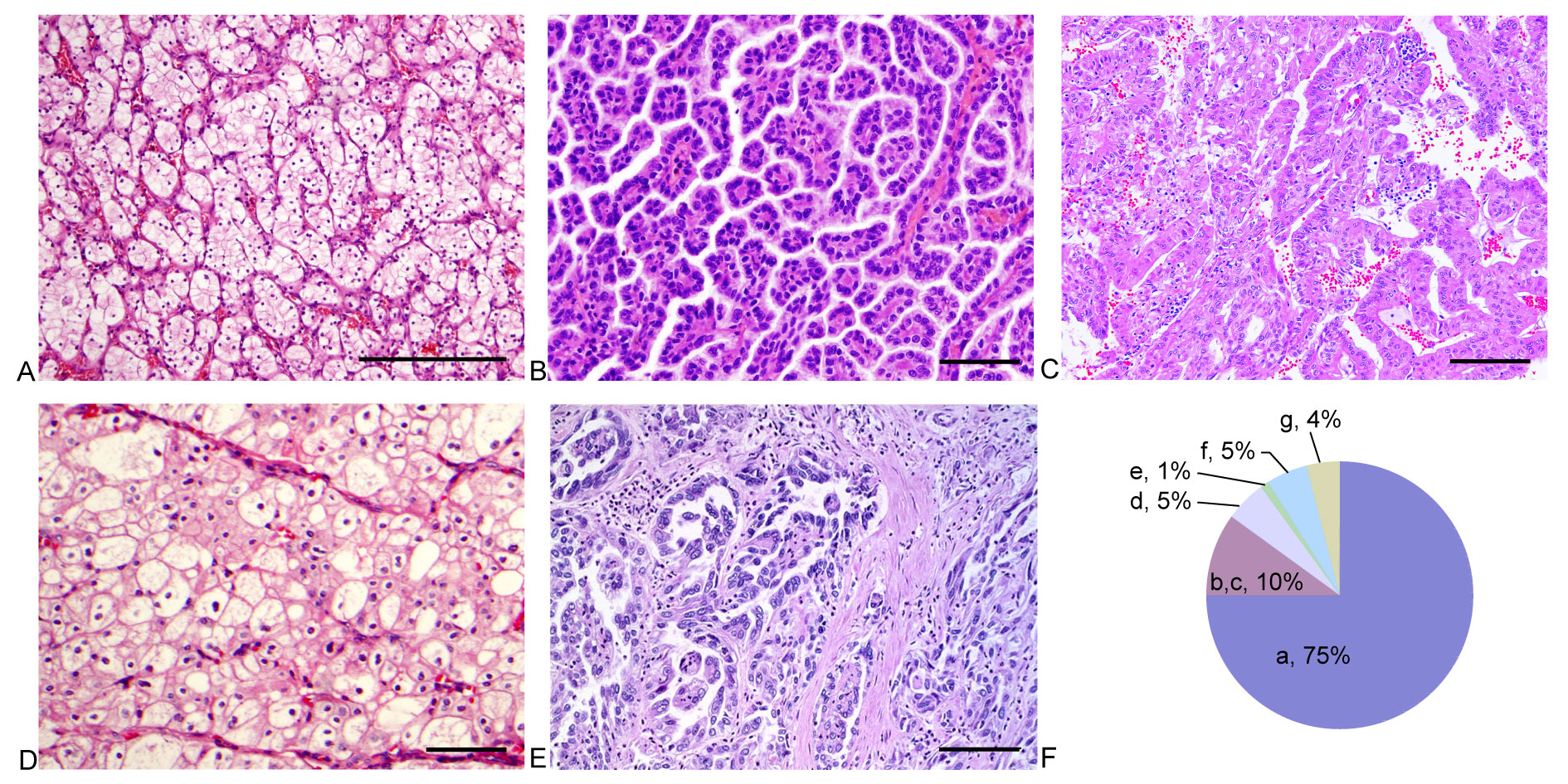
Figure 2. Pathological features for several main pathology types of renal cell carcinoma (RCC). A: clear cell renal cell carcinoma (ccRCC), low grade ccRCC with thin sinusoidal vessels and alveolar growth pattern; B: Type 1 papillary RCC, papillary RCC with a low grade nuclei, scanty cytoplasm and a single raw of tumor cells lining papillae; C: Type 2 papillary RCC, papillary RCC with a high grade nuclei, abundant eosinophilic cytoplasm and stratified/pseudostratified tumor cells lining papillae; D: Chromophobe RCC (chRCC), this tumor is characterized by distinct cell membrane, reisinoid nuclei and perinuclear clearing; E: Collecting duct carcinoma, this tumor shows tubular or tubulopapillary carcinoma with infiltrative growth, high nuclear grade and desmoplatic stroma; F: Pie chart for occurrence rate of several main pathology types of RCC (a: ccRCC; b: Type 1 papillary RCC; c: Type 2 papillary RCC; d: chRCC; e: Collecting duct carcinoma; f: Unclassified RCC; g: Other rare renal tumour). Scale bar = 100um.
| Table1: Classifiation of renal cell carcinoma by WTO in 2016 | |||||
| RCC subtype | Incidence(%) | Genetic | Histological characteristics | Gross appearance | Metastasis and Prognosis |
| Clear cell RCC | 75 | -3p,+5q22,-6q,-8p,-9p,-14 | Clear cytoplasm due to abundant lipid content of the cells; cells with eosinophilic cytoplasm | Solitary,rare multicentric or bilatera; typically golden yellow(necrosis, cystic degeneration, hemorrhage, calcification, ossification, extension into the renal vein and sarcomatoid change may occur) |
Aggressiveness according to grade, stage and sarcomatoid chang; most commonly metastasize hematogeneously via the vena cava primarily to the lung |
| Papillar RCC | 10 | +3q,+7,+8,+12,+16,+17,+20,-Y | Papillae and tubules formed by epithelial cells;macrophages;psammoma bodies;type 1(papillae covered by small cells with scanty cytoplasm) or type 2 (higher nuclear grade with eosinophilic cytoplasm and pseudostratified nuclei) | Multicentric, bilateral or solitary; frequently contain necrosis,sarcomatoid differentiation | Aggressiveness according to grade, stage and sarcomatoid chang |
| Chromophobe RCC | 5 |
-1, -2, -6, -10, -17, -21, hypodiploidy |
Large polygonal cells with reticulated cytoplasm and prominent cell membranes; irregular and multinucleated cells | Solitary; homogeneously gray or gray-brown | 10% mortality,distant metastasis into lung, liver(more frequently) and pancreas |
| Collecting duct carcinoma | 1 | -1q, -6p,-8p, -13q, -21q, -3p (rare) |
A tubulo-papillary architecture with a characteristic desmoplastic stroma reaction |
Solitary; located in the central region of the kidney and have a gray-white appearance with irregular borders | Extremely aggressive with frequent metastasis to regional lymph nodes, lung, liver, bone and adrenal glands |
| Renal medullary carcinoma | Rare | Unknown | Moderate to abundant eosinophilic cytoplasm with a hyperchromatic large nucleus containing a prominent nucleolus | Solitary; white, gray white, or tan white in color, firm, and infiltrative, with foci of necrosis | Metastases especially to the lung and bone; time from diagnosis to death averaged 3 months |
| Xp11 translocation carcinoma | Rare | t (X; 1) (p11.2; q21), t (X; 17) (p11.2; q25), Other |
Papillary architecture comprised of clear cells with voluminous clear to eosinophilic cytoplasm |
Solitary; unencapsulatedandis yellow, tan,and soft | Poor prognosis, presenting at advanced stages, frequently with lymph node metastasis at diagnosis |
| Multilocular cystic renal neoplasm of low malignant potential | Rare | VHL gene mutations | Clear cytoplasm, small dark nucle | Solitary, rare bilatera; complete cystic appearance without a solid tumoral component | No metastases |
| Succinate dehydrogenase-deficent renal carcinoma | Rare | SDH gene mutation | Clear cells or eosinophilic cells, cytoplasmic vacuoles | Solid brown, sometimes more red tumor,Cystic changes | Most of prognosis is good, case of sarcomatoid differentiation is less favorable |
| Mucinous tubular and spindle cell carcinoma | Rare | -1, -4, -6, -8, -13, -14,+7,+11,+16,+17 |
Low-grade renal epithelial neoplasms with tubular and spindle cell features and mucinous stroma |
Solitary; the cutsurfaceis yellow to tan brown,and the tumor is firm | Favorable prognosis and regional lymph nodes metastases |
| Tubulocystic RCC | Rare | +17,+7 | Cysts are lined by hobnail-like or big cells with huge nucleoli whose cytoplasm is eosinophilic and abundant | A multiple cystic sponge like aspect,and thin walls without vegetations | Only 4 of 70 reported cases showed metastasis to bone, liver, and lymph nodes |
| Acquired cystic disease-associated RCC | Rare | +1,+2,+3,+6,+7,+16,+17,+Y | The tumors are either solid or cystic or both and of papillary architecture. |
Most tumors were well-circumscribed, larger tumors with a thick fibrouscapsule;thesmalleroneswereilldefinedandirregular, which were filled with necrosis, coagulated blood, or transparent liquid |
Most tumours have indolent behaviour |
| Clear cell papillary RCC | Rare | -3p,+7,VHL, A358G, R120G mutations |
Low-grade clear epithelial cells arranged in tubules and papillae with a predominantly linear nuclear alignment away from the basement membrane |
Tan-white to yellow in color, well circumscribed, and well encapsulated | Indolent behaviour |
| RCC, unclassified | <5 | Unknown | Sarcomatoid morphology without recognizable epithelial elements, mucin production or unrecognizable cell types | Solitary; and the cut surface appeared grayish-red, with parts showing necrotic bleeding, resembling rotten fish specimens |
Highly aggressive biological behavior and poor clinical outcome |
| Papillary adenoma | Rare | Unknown | A tubulo-papillary architecture similar to cellular types 1 and 2 in papillar RCC | Solitary;well circumscribed, greyish or white lesion | Do not metastasize but should be cautious |
| Oncocytoma | Rare | Unknown | Densely granular eosinophilic cytoplasm and round and regular nuclei | Circumscribed, non-encapsulated, mahoganybrown or pale yellow with a central stellate scar | Benign tumor |
| VHL, von Hippel-Lindau disease tumor suppressor; SDH, succinate dehydrogenase; RCC, renal cell carcinoma. | |||||
There were two routes of renal cancer occurrence: a sporadic form and a hereditary form [46]. Over the years, numerous hereditary tumor syndromes with a tendency toward the development of RCC have been identified. Some mutant genes have been detected among patients with hereditary tumor syndromes, according to molecular analysis of their renal tumor tissues, such as the association of VHL, MET, FH, BHD and HRPT2 genes mutation with Von Hippel-Lindau disease, hereditary papillary RCC, hereditary leiomyomatosis and renal cell cancer syndromes (HLRCC), Birt-Hogg-Dubé (BHD) syndrome, and hyperparathyroidism-jaw tumor (HPT-JT) syndrome. These hereditary disease have been associated with various renal tumor pathological types. VHL disease, the most frequent familial renal cancer syndrome, is associated with ccRCC and with VHL wild type gene loss [47]. Hereditary papillary RCC syndrome and hereditary leiomyomatosis and renal cell cancer syndromes (HLRCC) are, respectively, associated with histological type 1 pRCC and type 2 pRCC [48, 49]. An activating mutation of the MET proto-oncogene and loss-of-function mutation in FH in the germ line are both respective features in patients. Renal tumors are one feature of these hereditary renal cancer symptoms, and there are many extrarenal organ manifestations, such as central nervous system (CNS) hemangioblastomas, pheochromocytoma and retinal angiomas, which also occur in patients with familial renal cancer syndrome together with renal tumors. Other hereditary renal cancer symptoms are summarized in Table 2 [2].
| Table2: Hereditary renal cell tumors | ||||
| Syndrome | Chromosome | Gene | Renal tumor | Other organ manifestations |
| Von Hippel-Lindau | 3p25-26 | VHL | Clear-cell RCC |
CNS haemangioblastomas; pheochromocytoma; retinal angiomas; pancreatic endocrine tumours; paragangliomas; cystadenomas of broad ligament or epididymis |
| Hereditary papillary RCC | 7q31-34 | c-MET | Type 1 papillary RCC | - |
| Hereditary leiomyomatosis and renal cell cancer syndromes | 1q42-43 | Fumarate hydratase | Type 2 papillary RCC | Leiomyomas of skin or uterus; uterine leiomyosarcomas |
| Hyperparathyreoidism-jaw tumor syndrome | 1q25 | HRPT2 |
Epithelial-stromal mixed tumors, papillary RCC |
Tumors of the parathyroidea; fibro-osseous jaw tumors |
| Birt-Hogg-Dubé syndrome | 17p11 | BHD |
Multiple chromophobe RCC, oncocytoma, papillary RCC |
Facial fibrofolliculoma; Pulmonal cysts; spontaneous pneumothorax |
| Tuberous Sclerosis |
9q34 or 16p13 |
TSC1 or TSC2 |
Multiple, bilateral angiomyolipomas, lymphangioleiomyo-matosis; Rare clear cell RCC |
Cardiac rhabdomyomas; neurological disorders or seizures; multiple skin fi ndings, including angiofi bromas, fi bromas, and nevi |
| Constitutional translocation chr.3 | 3p13-14 | - | Multiple, bilateral clear cell RCC | - |
| Familiary papillary thyroid carcinoma | 1q21 | - | Papillary RCC oncocytomas | Papillary thyroid carcinoma |
| RCC, renal cell carcinoma; CNS, central nerous system; TSC, Tuberous Sclerosis; MET, hepatocyte growth factor receptor; BHD, Birt-Hogg-Dubé; HRPT, hyperparathyroidism. | ||||
Various prognostic factors and models have been developed to evaluate the prognosis of RCC [50-53]. Thus far, a large amount of prognostic factors, including TNM stage, tumor factors, nuclear grade, histological type and clinical factors, have been discussed [54]. Although the accuracy of currently developed prognostic factors and staging systems remain controversial, the TNM system developed and maintained by the American Joint Committee on Cancer (AJCC) and the International Union Against Cancer (UICC) has been referred to extensively to evaluate the prognosis of renal cancer [55-57]. Like the abbreviation of the name in the TNM system, three key prognostic factors are associated with renal cancer: local extension of the primary tumor (T), involvement of regional lymph nodes (N), and presence of distant metastases (M). The current TNM system represents the eighth edition, and details of the edition are shown in Table 3 [57, 58].
Modification of this system has continued since the establishment of the TNM system in 1978 [59]. The major revision focuses on the local extension of the primary tumor (T). Various cut points (4.0, 4.5, 5.5 and 6.5 cm) have been established, even to explore the best cut point showing a significant difference in the prognosis of renal cancer. The cut point was defined as 2.5 cm in the 1978 edition of the TNM system, while the T2 category was expanded into four subcategories (T2a: > 2.5-5 cm; T2b: > 5-7.5 cm; T2c: > 7.5-10 cm; T2d: >10 cm) in the 1993 revised edition by AJCC. The current recommended 7 cm T1/T2 cut point was established in 1997 [60]. The current recommended 4 cm pT1a/pT1b cut point (reference value for partial nephrectomy) was established in 2002 according to the research results [61-63].
As evidence to define T3 and T4, it has been reported that renal tumors extending to perirenal fat, ipsilateral adrenal gland, renal sinus fat, kidney capsular and vena cava and renal vein thrombosis are associated with a worse prognosis, even if these conclusions remain controversial. Roberts et al. found that patients with pT1 RCC and pT3a RCC have the same recurrence-free survival rate [64]. Jeon et al. found that perirenal fat invasion has prognostic significance in patients with a tumor greater than 7 cm but less than or equal to 7 cm [65]. Adrenal gland involvement was noted in the TNM system in 1987. Initially, adrenal gland invasion or invasion into perirenal tissues (not beyond the Gerota fascia) was defined as T3a, and then it was defined as T4 due to the ipsilateral adrenal invasion with the same worse prognosis compared with tumor infiltration beyond the Gerota fascia [66, 67]. The renal sinus contains lymphatics and numerous large thin-walled tributaries of the main renal vein, and there is no fibrous capsule between the cortical tissue and the sinus, enabling RCC to gain access to the vein more readily than through the fibrous renal capsule [68]. The current TNM system regards renal sinus invasion as an important prognostic parameter in renal staging despite a few argumentative reports [69, 70]. Capsular invasion is more likely to be an independent prognostic factor in high size or grade tumors, but results have been inconsistent concerning the localization of T1 and T2 renal cancer [67, 71, 72]. Klaver et al. demonstrated that different levels of subdiaphragmatic tumor thrombus have significantly different cancer-specific survival, and they suggested a need to reclassify T3 of the 2002 version of the TNM system [73]. A retrospectively review of 1122 patients reported that patients with renal vein involvement have a 5-year survival rate of 43.2%; 37% have inferior vena cava (IVC) involvement below the diaphragm and 22% have caval involvement above the diaphragm [74]. Nevertheless, some studies have not found a significant difference in the survival rate between renal vein involvement and IVC invasion [75, 76]. The current version of the TNM system supports that renal vein, IVC involvement below the diaphragm and IVC involvement above the diaphragm should be classified as three different prognosis grades.
Regional lymph node involvement and distant metastasis have been demonstrated to be independent prognostic factors for a worse prognosis in the patients with renal cancer [77], providing evidence to define T3 and T4 in the current TNM system. In the 2002 TNM system, two grading levels for lymph nodes were distinguished: patients with one affected lymph node (N1) and those with multiple affected nodes (N2). The prognosis between N1 and N2 was controversial [78]. Terrone et al. demonstrated no statistically significant difference between pN1 and pN2 and supported that the involvement of 4 lymph nodes could be significantly differentiated from N1[56]. The clinical data clearly showed that distant metastasis had independent prognostic value and that bone or liver metastases were related to lung metastases in particular [79].
In addition, histological necrosis and microvascular tumor invasion [80, 81] were demonstrated to be independent prognostic factors for RCC. Fuhraman system has been frequently applied to grade RCC, but some studies show that the grading system is not suitable to grade chRCC and pRCC [82, 83]. Figure 3 evocatively shows the TNM stage of RCC and its relative management options.
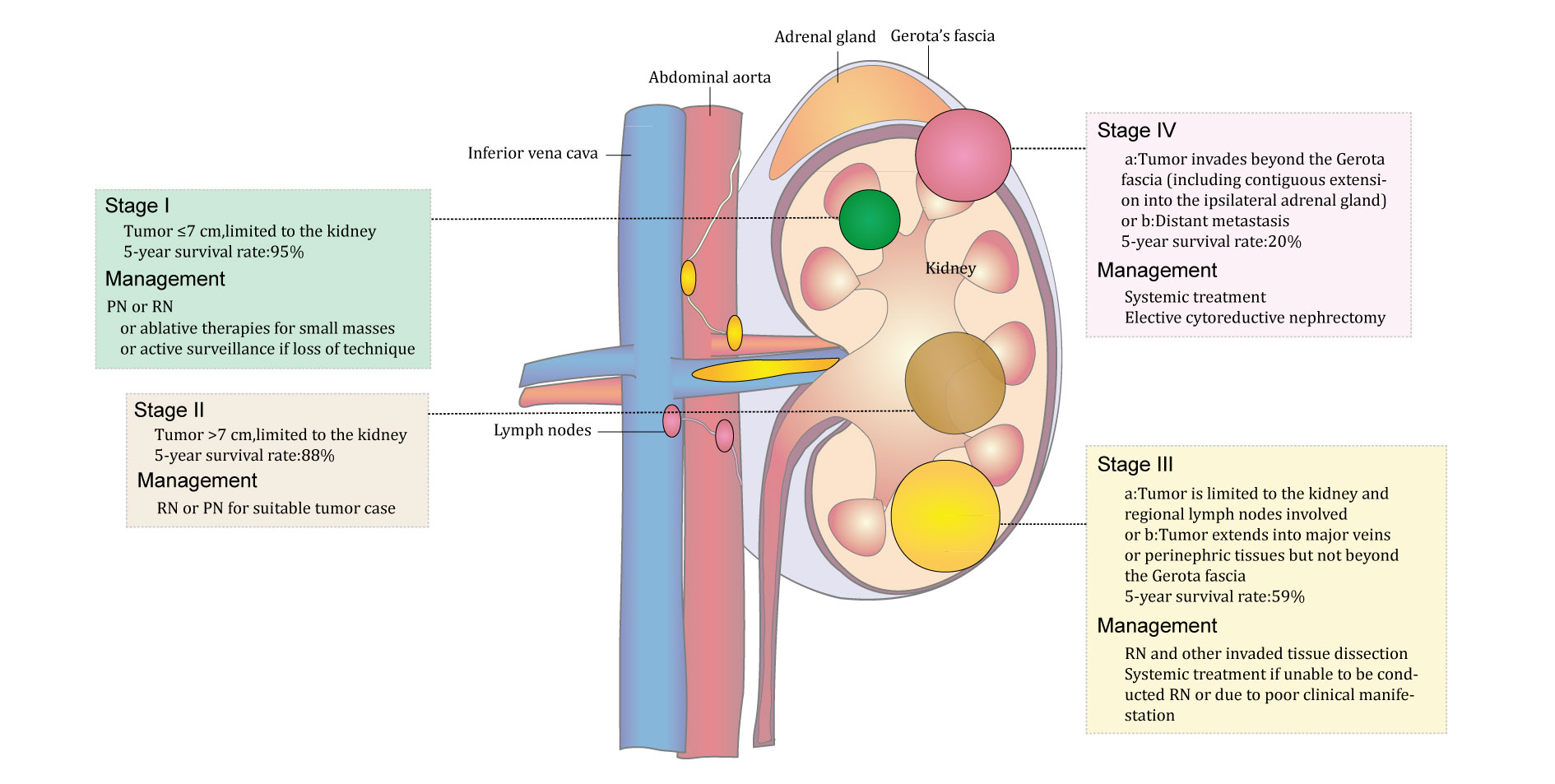 Figure 3. Schematic diagram of the stages of kidney cancer and relevant recommended treatment options.
Figure 3. Schematic diagram of the stages of kidney cancer and relevant recommended treatment options.
| Table3: TNM staging system for kidney cancer | |||||||
| Primary tumor(T) | Regional lymph nodes (N) | Distant metastasis (M) | |||||
| Tx | Primary tumor is unable to be evaluated | Nx | Regional lymph nodes cannot be assessed | M0 | No distant metastasis | ||
| T0 | No primary-tumor evidence | N0 | No regional lymph node metastasis | M1 | Distant metastasis | ||
| T1 | Tumor ≤7 cm,limited to the kidney | T1a | Tumor ≤ 4 cm | N1 | Metastasis in a single regional lymph node(s) | - | |
| T1b | Tumor > 4 cm | - | |||||
| T2 | Tumor >7 cm, limited to the kidney | T2a | Tumor ≤ 10 cm | ||||
| T2b | Tumor > 10 cm | ||||||
| T3 | Tumor extends into major veins or perinephric tissues but not into the ipsilateral adrenal gland and not beyond the Gerota fascia | T3a |
Tumor extends into the renal vein or its segmental (segmental vein) branches, or invasion of pelvicaliceal system or tumor invades perirenal and/or renal sinus fat but not beyond the Gerota fascia. |
||||
| T3b | Tumor grossly extends into the vena cava below the diaphragm | ||||||
| T3c | Tumor grossly extends into the vena cava above the diaphragm or invades the wall of the vena cava. | ||||||
| T4 | Tumor invades beyond the Gerota fascia (including contiguous extension into the ipsilateral adrenal gland) | ||||||
It has been reported that various genetic mutations contribute to RCC. Two main types of genetic mutations have been associated with ccRCC: mutation of the VHL gene and of some chromatin-modifier genes, including polybromo 1 (PBRM1), SET domain containing 2 (SETD2), BRCA1-associated protein-1 (BAP1), and lysine (K)-specific demethylase 5C (KDM5C) [84, 85]. The genetic alteration of Met is known to be related to pRCC. Nevertheless, not all cases of RCC exhibit these genetic alterations, and there are detectable rates of VHL or PBRM1 in ccRCC pathological specimens. Hakimi et al. found that the frequency of overall genetic mutation was 65% and VHL gene mutation was 49.2% in 185 ccRCC samples. They also reported that the alternation frequency rate of the chromatin-modifier genes PBRM1, BAP1, SETD2, and KDM5C in their 185 ccRCC samples was 29.2%, 5.9%, 7.6% and 7.6%, respectively [5]. The alteration frequency with one chromatin-modifier gene was 42% in ccRCC samples (Figure 4). Since its discovery, VHL has been deemed to be a very important genetic mutation that is significantly associated with ccRCC [4]. In fact, most patients with von Hippel–Lindau disease (a hereditary cancer syndrome) carry the mutation gene locus in the 25 subarea of the short arm of chromosome 3, with a high risk of renal cysts and clear cell kidney cancer. VHL gene inactivation in the renal epithelium can initiate ccRCC. VHL protein is a complex consisting of elongin B, elongin C, and cullin 2, and it functions to target hypoxia-inducible factors (HIFs) for ubiquitin-mediated degradation. Patients with von Hippel–Lindau disease inherit only one functional copy of the VHL gene and undergo a subsequent loss of heterozygosity (LOH) [86]. Loss of VHL normal function in the translation of VHL proteins will result in an accumulation of HIFs due to a loss of function in targeting HIFs for ubiquitin-mediated degradation. It has been demonstrated that the VHL protein complex functions in ubiquitin-mediated HIF degradation under normoxia conditions [86]. Because a proline residue of HIF-α must undergo hydroxylation for it to bind VHL protein [87, 88], the extent of hydroxylation depends on oxygen tension due to the gradual process of proline hydroxylases acting on HIF-α [89]. In addition, hydroxylation of an asparagine residue of the HIF-α amino acid sequence can also block its interaction with the transcriptional coactivator p300 [90]. The VHL complex fails to target HIFs for ubiquitin-mediated degradation, resulting in an accumulation of HIFs under hypoxia conditions. As transcription factors, HIFs consist of one HIF-α subunit (HIF1α, HIF2α, or HIF3α) and a member of the HIFβ family [89, 91]. HIF1-α or HIF2-α with HIF-β can bind to HIF response element (HER) in the cell nucleus to increase the mRNA levels of genes coding for vessel endothelial cell growth factors(VEGF), platelet derived growth factor(PDGFB), platelet derived growth factor 1(GLUT-1), transforming growth factor apha (TGF-α), erythropoietin, atypical protein kinase C and extracellular matrix protein, among others. The activating HIFs have effects on the angiogenesis, glycolysis and metastasis of cancer cells [34, 92]. Overall, the above two factors influence VHL gene loss mutations, and exposure of the VHL protein to hypoxia will result in a failure of HIFs to undergo ubiquitin-mediated degradation and thus their accumulation (Figure 5).
Recent studies have focused on several novel recurrent mutations in chromatin remodeling genes, mainly PBRM1, SETD2, BAP1, KDM5C, KDM6A and MLL2 [5, 84, 93-98]. These genes encode histone-modifying enzymes. SETD2, KDM5C, KMT2D and KDM6A were first identified in 2010, and PBRM1 and BAP1 were reported subsequently [84, 93]. Histone-lysine N-methyltransferase SETD2 (SETD2) is a histone H3 lysine 36 methyltransferase. Lysine-specific demethylase 5C (KDM5C) is a histone H3 lysine 4 demethylase. Histone lysine N- methyltransferase 2D (KMT2D namely, MLL2) is a histone H3 lysine 4 methyltransferase. Lysine-specific demethylase 6A (KDM6A) is a histone H3 lysine 27 demethylase [84]. Ubiquitin carboxyl-terminal hydrolase BAP1 (BAP1) is a deubiquitinase that targets the monoubiquitylation of lysine 119 on histone H2A [99]. Polybromo1 (PBRM1) is a component of the switch/sucrose nonfermentable (SWI/SNF) chromatin remodeling complex, which is involved in nucleosome repositioning [85]. DNA in eukaryotic cells that are not dividing is assembled around core histones (H2A,H2B,H3 and H4 family proteins) named the nucleosome. The nucleosome along with other protein complexes form a complex macromolecule called chromatin [100]. DNA transcription must be regulated by a series of actions of chromatin remodeling factors aided by a complex coding system of posttranslational modifications of the nucleosome within chromatin. The post-translational modifications include methylation, acetylation, phosphorylation, ubiquitylation, sumoylation, citrullination, and ADP ribosylation. Four types of chromatin remodelers have been identified, including SWI/SNF, imitation switch (ISWI), chromodomain helicase DNA-binding (CHD), and DNA helicase INO80 [101]. PBRM1, BAP1, SETD2, KDM5C and KDM6A have been reported to play important roles in posttranslational modification within chromatin. Thus, mutation of these genes and loss of these proteins will affect chromatin remodeling and DNA transcription. In general, chromatin-modifiers have two main functions in RCC, including canonical functions and noncanonical functions; the former is involved in DNA repair and transcriptional regulation, and the latter plays a role in the regulation of cytoskeletal dynamics and interferon signaling (Figure 5) [102]. It has been suggested that the loss of function of chromatin-modifiers, including PBRM1, BAP1, SETD2, and other related genes, is common in RCC, in which the mutation features of these genes can be considered as important codrivers of this disease. Nevertheless, the specific mechanism by which mutations of chromatin-modulating genes results in the pathogenesis of ccRCC is unknown. Interestingly, both VHL and these chromatin remodeling genes are all located on chromosome 3p, and ccRCC has been regarded as a disease of 3p loss, even if the links between VHL and chromatin-remodeling genes are unclear [6].
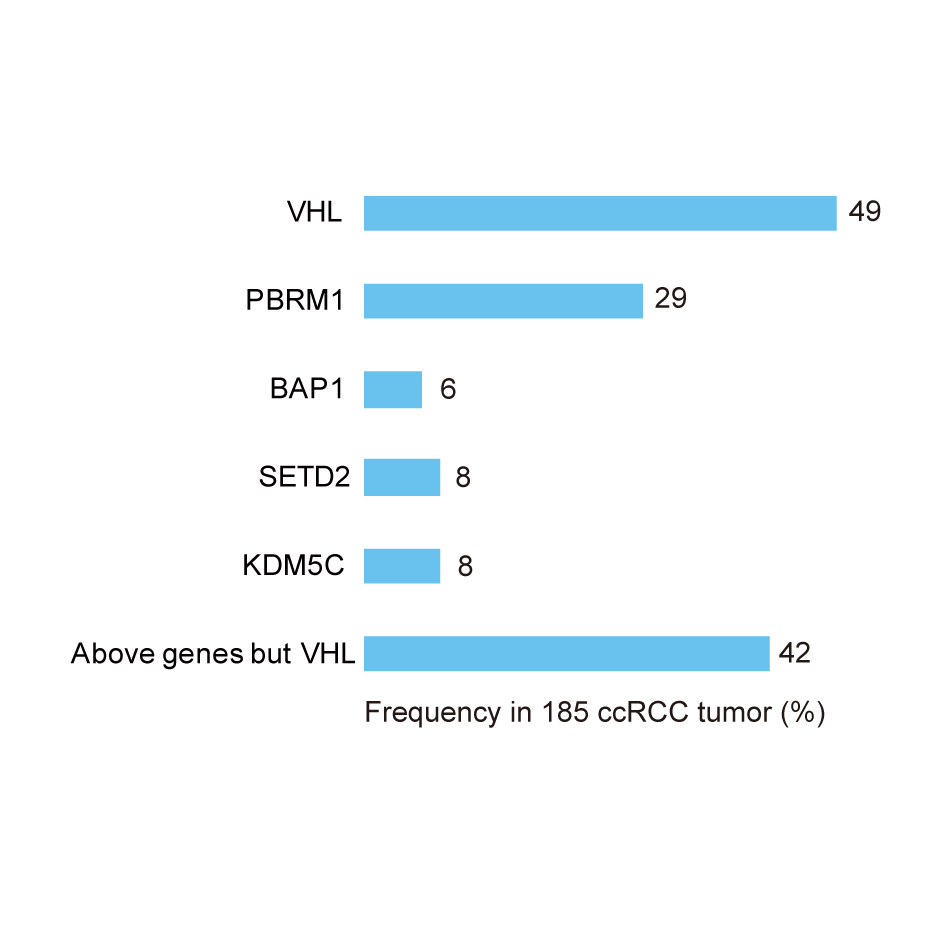 Figure 4. Mutation case frequency of VHL and chromatin-modifier genes (PBRM1, BAP1, SETD2, and KDM5C) in 185 ccRCC cases.
Figure 4. Mutation case frequency of VHL and chromatin-modifier genes (PBRM1, BAP1, SETD2, and KDM5C) in 185 ccRCC cases.

Figure 5. The genetic basis of ccRCC with VHL (located on 3p35) and chromatin-modifier gene (PBRM1, BAP1, SETD2, and KDM5C, mainly located on 3p21) mutations. VHL mutation: VHL protein is a complex consisting of elongin B, elongin C, and cullin 2 and functions to target HIFs for its ubiquitin-mediated degradation. VHL mutation and hypoxia exposure of VHL protein will result in a failure of HIFs to undergo ubiquitin-mediated degradation and accumulation. HIF1-α or HIF2-α with HIF-β can bind to HIF response element (HER) in the cell nucleus to increase mRNA levels coding for VEGF, PDGF, and CLUT1, among others. Chromatin-modifier gene mutation: Chromatin-modifiers have two main functions in ccRCC, including canonical functions and noncanonical functions. The former are involved in DNA repair and transcriptional regulation and the latter in the regulation of cytoskeletal dynamics and interferon signaling. Chromatin-modifier gene (PBRM1, BAP1, SETD2, and KDM5C) mutations and loss of their proteins will affect the above two main functions in ccRCC. ccRCC, clear cell renal cell carcinoma; VHL, von Hippel–Lindau disease tumor suppressor; HIF, hypoxia-inducible factors; VEGF, vessel endothelial cell growth factors; GLUT1, glucose transporter type 1; PDGF, platelet derived growth factor; +, Positive Activation.
The Met gene located on chromosome 7 was identified as a characteristic genetic mutation in pRCC [3]. Met encodes MET, a receptor tyrosine kinase that is capable of being activated by hepatocyte growth factor (HGF) [3]. Unlike the VHL mutation in ccRCC, the MET mutation is a duplication, resulting in an increased dose of the gene in pRCC cases [103]. The MET mutation is frequent in hereditary pRCC, where the MET receptor tyrosine kinase domain is subjected to auto-activating amino-acid-substitution, resulting in duplication of the MET gene [104]. Activation of MET will result in various changes in the biological signaling pathway. First, activating the phosphatidylinositol 3-kinase (PI3K) signaling pathway caused by the activation of MET will promote cell surface expression of nutrient transporters, which increases the uptake of amino acids, glucose and other nutrients [105]. This change promotes cell proliferation or tumor progression. Second, activation of MET can enable PI3K to activate mTORC2 with subsequent activation of HIF2-α [106]. Third, activation of MET can act on mTORC1 and subsequently on HIF1-α via activating AKT (positively activating) or the LKB1-AMPK and TSC1-TSC2 complex pathway (negatively activating)[107]. LKB1 (also referred to as STK11, serine threonine protein kinase 11) is the upstream kinase of 5' AMP-activated protein kinase (AMPK) (Figure 6) [106].
The TSC1-TSC2 complex is a heterodimer that consists of hamartin and tuberin, respectively, encoded by TSC1 and TSC2. The complex acts as a GTPase-activating protein toward Rheb. A Ras-family GTPase can activate mTORC1, but it action on Rheb is to inhibit mTOR activity[107]. Consequently, the effect of the MET mutation on the TSC1-TSC2 complex signaling pathway is to inhibit mTORC1 and HIF-α. Loss of TSC1 and TSC2 in RCC will result in inhibition of mTOR and HIF accumulation by activating mTORC1 and promoting tumor progression (Figure 6) [108]. Activating HIF2-α and HIF1-α have been suggested to affect angiogenesis, glycolysis and metastasis of cancer cells. Inhibitors of the PI3K, AKT and mTOR pathways presumably have anti-tumor effects [109]. In addition, various other gene mutation are involved in the occurrence and development of nonccRCC. In pRCC, in addition to the MET mutation, genetic mutations in NF2, SLC5A3, PNKD and CPQ have been found. TP53, PTEN, FAAH2, PDHB, PDXDC1 and ZNF765 gene mutations were identified in chRCC [110]. These mutant genes are more or less involved in the development and progression of the related renal cancer types.
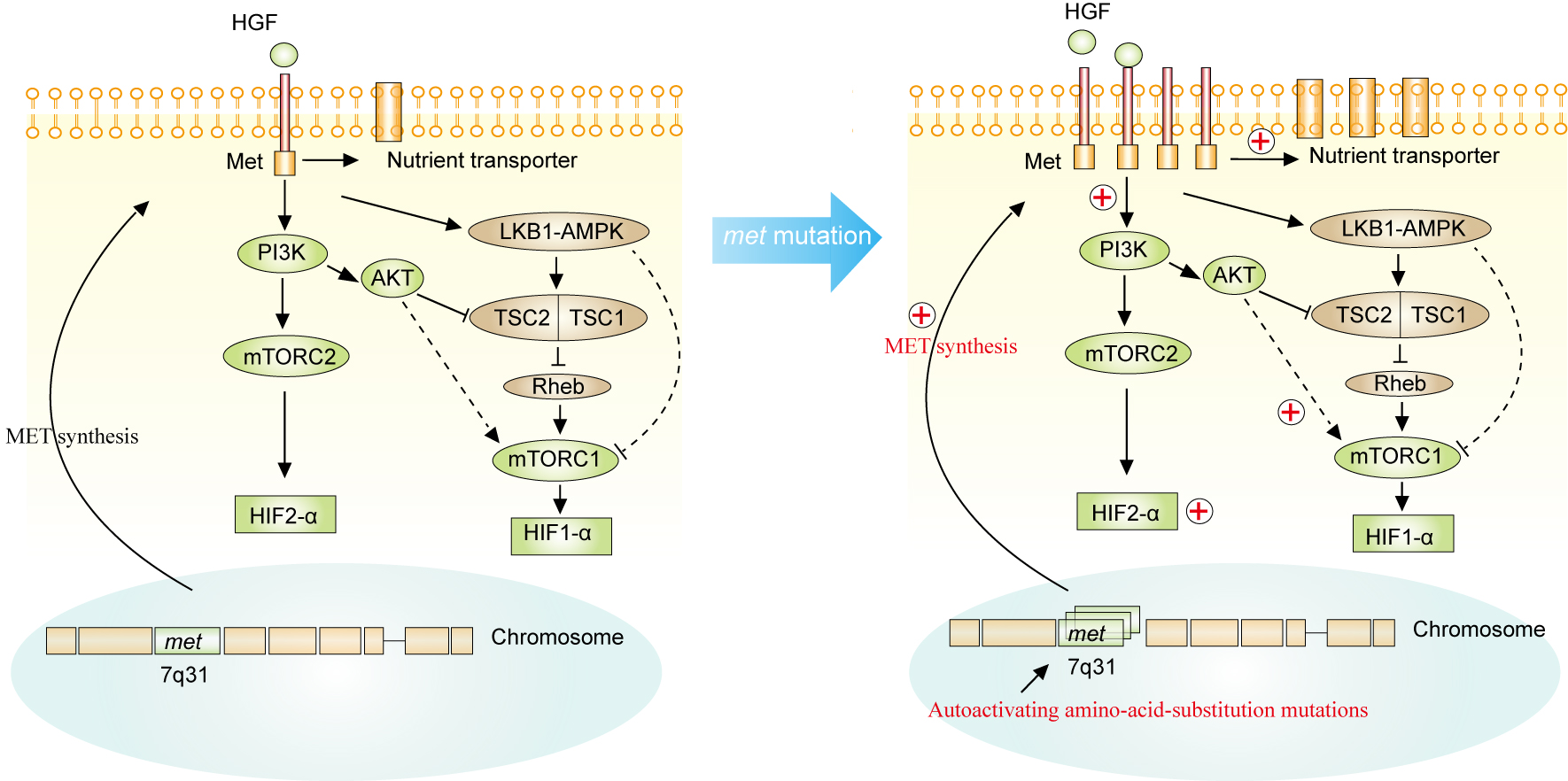
Figure 6. The genetic basis that MET (located on 7q31) of pRCC. MET mutation is duplicated, resulting in an increased dose of the gene in pRCC cases. Activation of MET will result in various changes in the signaling pathway biology. First, activation of the phosphatidylinositol 3-kinase (PI3K) signaling pathway by the activation of MET will promote cell surface expression of nutrient transporters, which increases the uptake of amino acids, glucose and other nutrients. Second, activation of MET can enable PI3K to activate mTORC2 with subsequent activation of HIF2-α. Third, activation of MET can act on mTORC1 and subsequently HIF1-α via activating AKT (activating) or the LKB1-AMPK and TSC1-TSC2 complex pathway (inhibiting). LKB1 (also referred to as STK11, serine threonine protein kinase 11) is the upstream kinase of 5' AMP-activated protein kinase (AMPK). The TSC1-TSC2 complex is a heterodimer consisting of hamartin and tuberin encoded, respectively, by TSC1 and TSC2. The complex acts as a GTPase-activating protein toward Rheb. A Ras-family GTPase can activate mTORC1, but it acts on Rheb is to inhibit mTOR activity. Akt, proto-oncogene c-Akt; AMPK, 5'-AMP-activated protein kinase; HGF, hepatocyte growth factor; HIF, hypoxia-inducible factor; LKB1, serine-threonine protein kinase 11; MET, hepatocyte growth factor receptor; mTOR, serine-threonine protein kinase mammalian target of rapamycin; PI3K, phosphatidylinositol 3-kinase; Rheb, GTP-binding protein Rheb; TSC, tuberous sclerosis complex; +, Positive Activation.
Traditionally, cancer has been regarded as a disease of uncontrolled cell proliferation mediated by oncogenes, while the metabolic disease concept of cancer has developed gradually as research has revealed metabolic pathway alterations in cancer [111-113]. Recently, kidney cancer may be considered a metabolic disease as a result of many of metabolic alterations in cancer, including several classical metabolic pathways [ 112]. These featured gene alterations in kidney cancer are directly linked to oncogenetic mutations [113, 114]. Such as loss of TSC1/2 leads to the Warburg effect and glutamine addiction via activating mTOR. VHL proteins can inhibit the Warburg effect via deactivation of HIF. LKB1 is associated with the upregulation of glycolysis and β-oxidation and downregulation of lipid synthesis via activation of AMPK [7]. Increasing GLUT-1 levels have been demonstrated in ccRCC samples compared with its normal control tissues, indicating that glucose uptake is increased in ccRCC [115]. Increased levels of glycolysis metabolites and enzymes such as phosphoglycerate kinase, hexokinase, pyruvate kinase 2, and LDH-A were identified in ccRCC cells and tissues in metabolomic, transcriptomic, proteomic, and transcriptomic research [116-118].
It has been suggested that the upregulation of glucose utilization for lactate fermentation is the sine qua non of the Warburg effect. Loss of VHL in ccRCC can increase HIF-1α, which is able to increase the expression of GLUT-1 to promote glucose uptake in cells [115]. Increased glucose can promote the TCA cycle, which is mediated by some rate-limiting enzymes including succinate dehydrogenase (SDH), fumarate hydratase (FH) and malate dehydrogenase. The TCA cycle was found to be significantly downregulated between succinate, fumarate and malate in kidney cancer compared with normal kidney tissues [116-119]. Succinate dehydrogenase (SDH) and/or fumarate hydratase (FH) deficiency can result in specific downregulation of the TCA cycle. SDH deficiency has been found to be related to familial paraganglioma and familial pheochromocytoma, and FH loss is associated with HLRCC [120, 121].
It has been reported that the PI3K-Akt-mTOR pathway is negatively regulated by PTEN and TSC1/2 and has a positive effect on mTOR, which increases the activity of anabolic pathways such as protein, lipid, and nucleotide synthesis in 28% of patients with ccRCC [94, 122]. Inactivation of the negative regulation of mTORC1, TSC1 and TSC2 results in an increased risk of ccRCC [122].
In addition, fatty acids and glutamine, arginine, and tryptophan metabolism abnormalities are associated with ccRCC. Fatty acids are elongated and desaturated by stearoyl-CoA desaturase (SCD1), which is increased in ccRCC tissues and is required for the growth and survival of ccRCC cells [123]. Studies have reported that glutamine utilization is increased in ccRCC compared with normal kidney tissues. Argininosuccinate synthase-1 (ASS1), the rate-limiting enzyme in tryptophan metabolism, is lost in tumor cells and highly expressed in normal proximal tubule cells [124]. Mutations in these key enzymes are associated with kidney cancer and thus may represent targets for therapeutic research in the clinic and lab.
The cytotoxic part of the immune system plays a vital role in the recognition and subsequent rejection of several different types of cancer. The cancer cell itself develops a system to escape the cytotoxicity of the immune system, notably the programmed death-1 (PD-1) pathway. The PD-1 pathway has been extensively valued by researchers in kidney cancer in recent years [125].
Normally, cytotoxic T lymphocytes (CD8+ T cells) recognize "foreign antigen" presented on the surface of cancer cells, which leads to the activation of cells and subsequent release of cytokines such as interferons, interleukin-2, and tumor necrosis factor. These cytokines are directly related to the death of the cancer cells. The cytokines and cytotoxic T lymphocytes can be regarded as therapeutic targets in ccRCC due to this type of cancer having features of immune T-cell infiltration. The PD-1 receptor is located in the cell membrane of CD8+ T cells, while programmed death ligand-1 (PD-L1) is presented on the surface of antigen-presenting cells and certain malignant cells, including RCC cells. The combination between the PD-1 receptor and its ligand PD-L1 has a biological effect on suppressing the cytotoxic immune system through inducing apoptosis of cytotoxic T lymphocytes. Thus, the effect of the PD-1 receptor and PD-L1 combination is anti-immune [126]. In several cancer types including ccRCC, PD-L1 is expressed on the cell surface, which can allow these cancer cells to reject the cytotoxic immune response via inducing apoptosis of cytotoxic T lymphocytes. Accordingly, some new agents, such as anti-PD-1 or anti-PD-L1 antibodies that block PD-1 and PD-L1, have been developed to treat cancer including ccRCC due to their effect on inhibiting the anti-immune response in cancer with PD-1/PD-L1 (Figure 7). PD-1 blocking antibodies include nivolumab and pembrolizumab, and PD-L1-blocking monoclonal antibodies include atezolizumab and avelumab [127]. Another protein receptor, CTLA-4, is present on the surface of cytotoxic T lymphocytes and exhibits an anti-immune function similar to the PD-1/PD-L1 pathway. The corresponding inhibitors, including ipilimumab, are under investigation with promising result for the treatment of metastatic RCC [126, 128].

Figure 7. The programmed death-1 receptor (PD-1) pathway. PD-1 participating in T-cell binding to PD-L1 (programmed death-1 ligand) expressed on cancer cell is able to promote apoptosis of T-cells (CD8+). Anti-PD-1 and anti-PD-L1 antibodies are able to block the combination PD-1 and PD-L1 to inhibit apoptosis of T-cells (CD8+) and result in lysis of tumor cells. MHC, main histocompatibility complex; PD-1, programmed death-1 receptor; PD-L1, programmed death-1 ligand; TCR, T cell receptor.
Surgery
Surgical treatment remains the first considerable cure for patients with surgically resectable RCC. Different surgical procedures (elective partial nephrectomy: PN, radical nephrectomy: RN, and focal therapy) are recommended to treat RCC of different clinical stages [129, 130]. PN is to remove the primary tumor while preserving those renal tissues with normal function. Traditional radical nephrectomy (RN) is to remove the tissues of these organs, including the kidney, perirenal fat tissue, adrenal gland and regional lymph nodes. Current guideline recommends PN as the standard management for clinical T1a (cT1a) renal tumors (≤4.0 cm) [131]. RN is preferred for clinical T1b (cT1b) renal tumors ( > 4.0 cm to < 7 cm), while PN is favored over RN in the patients if technically feasible [132]. Nevertheless, RN is known to treat cT2 renal tumor, but there is evidence that some cases of T2 renal tumor may be selective for treatment with PN[133]. PN and RN mainly focus on these local renal tumors, and for metastatic RCC, cytoreductive nephrectomy with postoperative adjuvant therapy is usually applied. Small renal masses ( < 3 cm) can be treated with focal therapy using percutaneous, laparoscopic or open approaches. Cryoablation of the local renal mass using liquid argon or nitrogen with different freeze-thaw cycles results in the denaturation and destruction of the tumor tissue. Whole kidney removal (such as open RN) was regarded to have a better effect in terms of oncological outcome in the past, but it results in the loss of some health kidney tissue[129]. A recent Cochrane review identified that PN may be associated with a decreased time-to-death of any cause, even though it is unable to reduce surgery-related mortality, cancer-specific survival and time-to-recurrence [134]. How to select the suitable case of renal tumor remains vital for clinicians to treat renal cancer.
Minimally invasive surgical technology has developed rapidly, and there are various surgical techniques used in the clinic including open, laparoscopic and robotic PN and RN. Jame et al. compared the therapeutic effects for patients with local renal tumors undergoing three different nephrectomy surgical procedures, including open, laparoscopic and robotic PN. They found that robotic PN was more effective than laparoscopic PN as a minimally invasive approach due to its more significant centralization compared with the more uniform distribution of open PN and polarization of laparoscopic PN surgeon experience [135, 136]. The application of robotic and laparoscopic PN is able to decrease the risk of blood transfusion and length of stay compared with open PN. In addition, robotic PN can reduce the risk of inpatient complications. Nevertheless, robotic PN significantly increases hospital costs [135]. A review by Ng et al. showed that laparoscopic PN reduced renal ischemia time and exhibited other functional outcomes equivalent to open PN [137]. An updated meta-analysis has shown that robotic PN favors patients compared with laparoscopic RN in terms of perioperative outcomes of estimated blood loss (EBL), length of stay (LOS) and warm ischemia time (WIT) [138]. Considering cost and effect, laparoscopic PN seems to be more suitable to the patient's burden. Minimally invasive nephrectomy, including robotic or laparoscopic RN, has been demonstrated to be more effective to treat advanced RCC in terms of reduction of estimated blood loss (EBL), transfusion rate and length of stay than traditional open RN [139]. Commonly, PN is more technically challenging to manipulate than RN. Researchers have also compared the effects of three different operative techniques on RN: open, laparoscopic and robotic RN. While they found that robotic-assisted RN increased medical costs and did not improve patient morbidity compared with laparoscopic RN [140], they did not recommend robotic-assisted RN to perform pure RN if laparoscopic RN was manipulated by the surgeon proficiently [140].
Adjuvant therapy
Adjuvant therapy of RCC consists of immunotherapy and targeted therapy. Early immunotherapy treatment strategies indicated that cytokines, including interferon-α (INF-α) and high-dose IL-2, be applied to treat metastatic RCC as a standard of care from the 1990s when anti-VEGF agents (sunitinib) were not approved by the FDA. Nevertheless, both cytokines were limited by their availability range and substantial toxicity (especially high-dose IL-2) [141]. New immunotherapy agents for cancers continue to be developed by researchers. Active specific immunotherapy has been shown to prevent tumors in in vitro studies and small trials as early as the 1990s. A randomized study was designed to identify the effectiveness of intradermal injections of a vaccine produced from 107 autologous irradiated tumor cells subsequently admixed with Bacillus Calmette Guerin (BCG) in postnephrectomy patients. The result was disappointing due to the inferior 5-year progression free survival and overall survival rate in the vaccine group compared with the placebo group [142]. Another similar study using an autologous tumor lysate vaccine (incubated with IFN-γ and tocopherol acetate but without addition of any cytokines, bacterial, or viral adjuvants) showed an improved 5-year survival probability in the patients treated with the vaccine [143]. Due to the controversial results and complexity of vaccine manufacture, vaccine application in the treatment of RCC is challenging. A new therapeutic era for cancer management is represented by immune check point inhibitors. The most prevalent studies on immune check point inhibitors have investigated PD-1/PD-L1 and CTLA4, which have been described in the above text. The combination of PD-1 expressed in T cells and its ligand (PD-L1) expressed in cancer cells has anti-tumor immune effects. Anti-PD-1 and anti-PD-L1 antibodies can inhibit the anti-tumor immune effects of PD-1 and PD-L1 and increase the anti-tumor effect of T cells. Due to the similar anti-tumor effect of CTLA4, CTLA4 inhibitors can increase its anti-tumor function. Nivolumab and pembrolizumab are humanized anti-PD-1 monoclonal antibodies that have been newly developed in 2015 to block the combination of PD-1 and PD-L1. A randomized Phase 3 study identified that nivolumab can be the standard of care in previously treated patients with advanced RCC [144]. In addition, overall survival was longer with fewer grade 3 or 4 scores in renal cancer patients treated with nivolumab than everolimus [145]. A recent study has shown that treatment with a combination of axitinib plus pembrolizumab is tolerable, suggesting a promising anti-tumor activity in patients with advanced renal cell cancer [146]. Atezolizumab and avelumab are PD-L1 inhibitors that have studied in combination with anti-VEGF agents in a recent clinical trial and demonstrated some improvement of progression-free survival (PFS) in patients with renal carcinoma [147]. Ipilimumab is a CTLA4 inhibitor that was approved by the FDA for the treatment of melanoma in 2011 [148]. A study by Motzer et al. suggested that treatment with nivolumab plus ipilimumab in patients with advanced RCC was superior to sunitinib treatment alone [149]. A recent review has shown that checkpoint inhibitors show promising results, suggesting that new immune modulatory treatments will dramatically change the current management situation for RCC [8].
From the basic biology of several main types of RCC, two types of targeted therapeutic agents have been developed for extensive use in the clinic, including anti-VEGF agents and mTOR inhibitors. Since 2015, the anti-VEGF agents that have been demonstrated to be effective and approved by the FDA are sunitinib and sorafenib [150, 151]. Sunitinib is more effective to increase PFS than IFN-α in patients with metastatic renal-cell cancer [150]. Sorafenib has demonstrated more adverse events compared with placebo, even if it is effective for improving FPS [151]. In 2007, an anti-VEGF agent, bevacizumab, plus IFN-α combination treatment as a first-line treatment was demonstrated to significantly improve PFS in metastatic RCC [152]. Recently, more anti-VEGF agents targeting downstream mediators of the HIF activation pathway have been approved by the FDA for the treatment of advanced or metastatic RCC, such as pazopanib, axitinib, cabozantinib, lenvatinib and nivolumab. Generally, a single application of sunitinib, pazopanib and the combination of INF-α and bevacizumab are recommended as first-line options; axitinib and cabozantinib are approved as second-line options [153]. These agents are almost all VEGFR tyrosine kinase inhibitors (TKIs) that can inhibit various VEGFRs and other relative growth regulatory receptors [154]. mTOR inhibitors are able to block the mTOR signal pathway involved in the regulation of cell growth, proliferation, metabolism and angiogenesis. Following the previous introduction of the genetic mechanism of RCC, the mTOR signal pathway was also shown to play a role in the development and progression of RCC. Two representative mTOR inhibitors are everolimus and tesirolimus. mTOR inhibitors were approved as second-line and first-line treatment and for metastatic RCC [155]. In a comparison of anti-VEGF agents and mTOR inhibitors, a systematic review demonstrated that sunitinib might be more effective than everolimus for nonccRCC even if the mTOR signal pathway has been identified to be featured in nonccRCC [156]. These developed targeting therapy agents like anti-VEGF agents suffer from side effects, including diarrhea, hypertension, fatigue, and nausea. Anti-mTOR inhibitors have side effects hyperglycemia, hyperlipidemia, and hypercholesterolemia. More effective targeting therapeutic agents with fewer side effects remain be exploited in the future. Figure 8 shows the therapeutic evolution of immunotherapy and targeted therapies for RCC.
Overall, according to current guidelines for RCC, PN and RN, respectively, are recommended to treat smaller local RCC (less than 7 cm) and larger local RCC (more than 7 cm). Adjuvant therapy is applied in patients with more progressive RCC such as Stage III and Stage IV RCC. Surveillance strategies and cryoablation are applied to treat the local renal mass if a PN is difficult to perform [130]. The management options for RCC of different stages are represented in Figure 3.

Figure 8. Therapeutic evolution of immunotherapies and targeted therapies in renal cell carcinoma.
The global incidence of kidney cancer is increasing rapidly and is mainly distributed in developed countries. Various factors have an effect on RCC, such as race, age, sex, smoking and obesity, hypertension and the use of antihypertensive medications, acquired renal cystic disease, diabetes and urinary tract infection, nutritional factors and diet, occupation, and genetic factors. Traditionally, VHL and MET genetic mutations have been the genetic features of ccRCC and pRCC, respectively. It was recently noted by researchers that genetic mutations occur in some chromatin-modifier genes, including polybromo 1 (PBRM1), SET domain containing 2 (SETD2), BRCA1-associated protein-1 (BAP1), and lysine (K)-specific demethylase 5C (KDM5C). The metabolic disease concept in renal cancer has been noted by the researchers worldwide. The PD-1 pathway has been valued by researchers in kidney cancer in recent years, and new agents, such as anti-PD-1 monoclonal antibodies (nivolumab and pembrolizumab) and CTLA4 inhibitors (ipilimumab) were approved to treat advanced RCC. Partial nephrectomy (PN) and radical nephrectomy (RN) remain the treatment standards for T1 and T2 stage local RCC, respectively. PN can be used for T2 stage RCC in suitable cases. Even if targeted therapy such as anti-VEGF and anti-mTOR pathway agents are recommended as first-line and second-line treatments for advanced RCC, their effectiveness and side effects remain noteworthy and necessitate further studies. For translational research of ccRCC, regulation of the mutations in chromatin-modifier genes in ccRCC, the association between these gene mutation and VHL mutations, the affected metabolic pathways and the connections among all the genetic mutation merit investigation. In addition, the future will probably represent an era of research and application of immune check point inhibitor treatment in RCC.
-
Wong MCS, Goggins WB, Yip BHK, Fung FDH, Leung C, Fang Y, Wong SYS, Ng CF: Incidence and mortality of kidney cancer: temporal patterns and global trends in 39 countries. Sci Rep 2017, 7(1):15698. [Pubmed]
-
Moch H: An overview of renal cell cancer: pathology and genetics. Semin Cancer Biol 2013, 23(1):3-9. [Pubmed]
-
Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P, Scherer SW, Zhuang Z, Lubensky I, Dean M et al: Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet 1997, 16(1):68-73. [Pubmed]
-
Gnarra JR, Tory K, Weng Y, Schmidt L, Wei MH, Li H, Latif F, Liu S, Chen F, Duh FM et al: Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet 1994, 7(1):85-90. [Pubmed]
-
Hakimi AA, Chen YB, Wren J, Gonen M, Abdel-Wahab O, Heguy A, Liu H, Takeda S, Tickoo SK, Reuter VE et al: Clinical and pathologic impact of select chromatin-modulating tumor suppressors in clear cell renal cell carcinoma. Eur Urol 2013, 63(5):848-854. [Pubmed]
-
Hakimi AA, Pham CG, Hsieh JJ: A clear picture of renal cell carcinoma. Nat Genet 2013, 45(8):849-850. [Pubmed]
-
Wettersten HI, Aboud OA, Lara PN, Jr., Weiss RH: Metabolic reprogramming in clear cell renal cell carcinoma. Nat Rev Nephrol 2017, 13(7):410-419. [Pubmed]
-
Flippot R, Escudier B, Albiges L: Immune Checkpoint Inhibitors: Toward New Paradigms in Renal Cell Carcinoma. Drugs 2018, 78(14):1443-1457. [Pubmed]
-
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM: Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010, 127(12):2893-2917. [Pubmed]
-
Dy GW, Gore JL, Forouzanfar MH, Naghavi M, Fitzmaurice C: Global Burden of Urologic Cancers, 1990-2013. Eur Urol 2017, 71(3):437-446. [Pubmed]
-
King SC, Pollack LA, Li J, King JB, Master VA: Continued increase in incidence of renal cell carcinoma, especially in young patients and high grade disease: United States 2001 to 2010. J Urol 2014, 191(6):1665-1670. [Pubmed]
-
Znaor A, Lortet-Tieulent J, Laversanne M, Jemal A, Bray F: International variations and trends in renal cell carcinoma incidence and mortality. Eur Urol 2015, 67(3):519-530. [Pubmed]
-
Karim-Kos HE, de Vries E, Soerjomataram I, Lemmens V, Siesling S, Coebergh JW: Recent trends of cancer in Europe: a combined approach of incidence, survival and mortality for 17 cancer sites since the 1990s. Eur J Cancer 2008, 44(10):1345-1389. [Pubmed]
-
Chow WH, Devesa SS: Contemporary epidemiology of renal cell cancer. Cancer J 2008, 14(5):288-301. [Pubmed]
-
Mortality GBD, Causes of Death C: Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385(9963):117-171. [Pubmed]
-
Global Burden of Disease Cancer C, Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M, MacIntyre MF, Allen C, Hansen G, Woodbrook R et al: The Global Burden of Cancer 2013. JAMA Oncol 2015, 1(4):505-527. [Pubmed]
-
Ljungberg B, Campbell SC, Choi HY, Jacqmin D, Lee JE, Weikert S, Kiemeney LA: The epidemiology of renal cell carcinoma. Eur Urol 2011, 60(4):615-621. [Pubmed]
-
Cumberbatch MG, Rota M, Catto JW, La Vecchia C: The Role of Tobacco Smoke in Bladder and Kidney Carcinogenesis: A Comparison of Exposures and Meta-analysis of Incidence and Mortality Risks. Eur Urol 2016, 70(3):458-466. [Pubmed]
-
Callahan CL, Hofmann JN, Corley DA, Zhao WK, Shuch B, Chow WH, Purdue MP: Obesity and renal cell carcinoma risk by histologic subtype: A nested case-control study and meta-analysis. Cancer Epidemiol 2018, 56:31-37. [Pubmed]
-
Sanfilippo KM, McTigue KM, Fidler CJ, Neaton JD, Chang Y, Fried LF, Liu S, Kuller LH: Hypertension and obesity and the risk of kidney cancer in 2 large cohorts of US men and women. Hypertension 2014, 63(5):934-941. [Pubmed]
-
Chow WH, Gridley G, Fraumeni JF, Jr., Jarvholm B: Obesity, hypertension, and the risk of kidney cancer in men. N Engl J Med 2000, 343(18):1305-1311. [Pubmed]
-
Hidayat K, Du X, Zou SY, Shi BM: Blood pressure and kidney cancer risk: meta-analysis of prospective studies. J Hypertens 2017, 35(7):1333-1344. [Pubmed]
-
Port FK, Ragheb NE, Schwartz AG, Hawthorne VM: Neoplasms in dialysis patients: a population-based study. Am J Kidney Dis 1989, 14(2):119-123. [Pubmed]
-
Nouh MA, Kuroda N, Yamashita M, Hayashida Y, Yano T, Minakuchi J, Taniguchi S, Nomura I, Inui M, Sugimoto M et al: Renal cell carcinoma in patients with end-stage renal disease: relationship between histological type and duration of dialysis. BJU Int 2010, 105(5):620-627. [Pubmed]
-
Zucchetto A, Dal Maso L, Tavani A, Montella M, Ramazzotti V, Talamini R, Canzonieri V, Garbeglio A, Negri E, Franceschi S et al: History of treated hypertension and diabetes mellitus and risk of renal cell cancer. Ann Oncol 2007, 18(3):596-600. [Pubmed]
-
Parker AS, Cerhan JR, Lynch CF, Leibovich BC, Cantor KP: History of urinary tract infection and risk of renal cell carcinoma. Am J Epidemiol 2004, 159(1):42-48. [Pubmed]
-
Chow WH, Lindblad P, Gridley G, Nyren O, McLaughlin JK, Linet MS, Pennello GA, Adami HO, Fraumeni JF, Jr.: Risk of urinary tract cancers following kidney or ureter stones. J Natl Cancer Inst 1997, 89(19):1453-1457. [Pubmed]
-
Lee JE, Spiegelman D, Hunter DJ, Albanes D, Bernstein L, van den Brandt PA, Buring JE, Cho E, English DR, Freudenheim JL et al: Fat, protein, and meat consumption and renal cell cancer risk: a pooled analysis of 13 prospective studies. J Natl Cancer Inst 2008, 100(23):1695-1706. [Pubmed]
-
Song DY, Song S, Song Y, Lee JE: Alcohol intake and renal cell cancer risk: a meta-analysis. Br J Cancer 2012, 106(11):1881-1890. [Pubmed]
-
Zhang S, Jia Z, Yan Z, Yang J: Consumption of fruits and vegetables and risk of renal cell carcinoma: a meta-analysis of observational studies. Oncotarget 2017, 8(17):27892-27903. [Pubmed]
-
Karami S, Lan Q, Rothman N, Stewart PA, Lee KM, Vermeulen R, Moore LE: Occupational trichloroethylene exposure and kidney cancer risk: a meta-analysis. Occup Environ Med 2012, 69(12):858-867. [Pubmed]
-
Mundt KA, Birk T, Burch MT: Critical review of the epidemiological literature on occupational exposure to perchloroethylene and cancer. Int Arch Occup Environ Health 2003, 76(7):473-491. [Pubmed]
-
Cohen D, Zhou M: Molecular genetics of familial renal cell carcinoma syndromes. Clin Lab Med 2005, 25(2):259-277. [Pubmed]
-
Melendez-Rodriguez F, Roche O, Sanchez-Prieto R, Aragones J: Hypoxia-Inducible Factor 2-Dependent Pathways Driving Von Hippel-Lindau-Deficient Renal Cancer. Front Oncol 2018, 8:214. [Pubmed]
-
Courthod G, Tucci M, Di Maio M, Scagliotti GV: Papillary renal cell carcinoma: A review of the current therapeutic landscape. Crit Rev Oncol Hematol 2015, 96(1):100-112. [Pubmed]
-
Kovacs G, Akhtar M, Beckwith BJ, Bugert P, Cooper CS, Delahunt B, Eble JN, Fleming S, Ljungberg B, Medeiros LJ et al: The Heidelberg classification of renal cell tumours. J Pathol 1997, 183(2):131-133. [Pubmed]
-
Lopez-Beltran A, Scarpelli M, Montironi R, Kirkali Z: 2004 WHO classification of the renal tumors of the adults. Eur Urol 2006, 49(5):798-805. [Pubmed]
-
Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM: The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur Urol 2016, 70(1):93-105. [Pubmed]
-
Srigley JR, Delahunt B, Eble JN, Egevad L, Epstein JI, Grignon D, Hes O, Moch H, Montironi R, Tickoo SK et al: The International Society of Urological Pathology (ISUP) Vancouver Classification of Renal Neoplasia. Am J Surg Pathol 2013, 37(10):1469-1489. [Pubmed]
-
Chen WJ, Pan CC, Shen SH, Chung HJ, Lin CC, Lin ATL, Chang YH: Clear cell papillary renal cell carcinoma - An indolent subtype of renal tumor. J Chin Med Assoc 2018. [Pubmed]
-
Comperat E, Varinot J: Classification of Adult Renal Tumors: An Update. Semin Ultrasound CT MR 2017, 38(1):2-9. [Pubmed]
-
Moch H, Gasser T, Amin MB, Torhorst J, Sauter G, Mihatsch MJ: Prognostic utility of the recently recommended histologic classification and revised TNM staging system of renal cell carcinoma: a Swiss experience with 588 tumors. Cancer 2000, 89(3):604-614. [Pubmed]
-
Kovacs G: Papillary renal cell carcinoma. A morphologic and cytogenetic study of 11 cases. Am J Pathol 1989, 134(1):27-34. [Pubmed]
-
Nordqvist SR, Fidler WJ, Jr., Woodruff JM, Lewis JL, Jr.: Clear cell adenocarcinoma of the cervix and vagina. A clinicopathologic study of 21 cases with and without a history of maternal ingestion of estrogens. Cancer 1976, 37(2):858-871. [Pubmed]
-
Thoenes W, Storkel S, Rumpelt HJ, Moll R, Baum HP, Werner S: Chromophobe cell renal carcinoma and its variants--a report on 32 cases. J Pathol 1988, 155(4):277-287. [Pubmed]
-
Linehan WM, Gnarra JR, Lerman MI, Latif F, Zbar B: Genetic basis of renal cell cancer. Important Adv Oncol 1993:47-70. [Pubmed]
-
Neumann HP, Bender BU, Berger DP, Laubenberger J, Schultze-Seemann W, Wetterauer U, Ferstl FJ, Herbst EW, Schwarzkopf G, Hes FJ et al: Prevalence, morphology and biology of renal cell carcinoma in von Hippel-Lindau disease compared to sporadic renal cell carcinoma. J Urol 1998, 160(4):1248-1254. [Pubmed]
-
Salvi A, Marchina E, Benetti A, Grigolato P, De Petro G, Barlati S: Germline and somatic c-met mutations in multifocal/bilateral and sporadic papillary renal carcinomas of selected patients. Int J Oncol 2008, 33(2):271-276. [Pubmed]
-
Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, Simonds WF, Gillanders EM, Kennedy AM, Chen JD et al: HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet 2002, 32(4):676-680. [Pubmed]
-
Zigeuner R, Hutterer G, Chromecki T, Imamovic A, Kampel-Kettner K, Rehak P, Langner C, Pummer K: External validation of the Mayo Clinic stage, size, grade, and necrosis (SSIGN) score for clear-cell renal cell carcinoma in a single European centre applying routine pathology. Eur Urol 2010, 57(1):102-109. [Pubmed]
-
Zisman A, Pantuck AJ, Wieder J, Chao DH, Dorey F, Said JW, deKernion JB, Figlin RA, Belldegrun AS: Risk group assessment and clinical outcome algorithm to predict the natural history of patients with surgically resected renal cell carcinoma. J Clin Oncol 2002, 20(23):4559-4566. [Pubmed]
-
Kattan MW, Reuter V, Motzer RJ, Katz J, Russo P: A postoperative prognostic nomogram for renal cell carcinoma. J Urol 2001, 166(1):63-67. [Pubmed]
-
Sorbellini M, Kattan MW, Snyder ME, Reuter V, Motzer R, Goetzl M, McKiernan J, Russo P: A postoperative prognostic nomogram predicting recurrence for patients with conventional clear cell renal cell carcinoma. J Urol 2005, 173(1):48-51. [Pubmed]
-
Furniss D, Harnden P, Ali N, Royston P, Eisen T, Oliver RT, Hancock BW, National Cancer Research Institute Renal Clinical Studies G: Prognostic factors for renal cell carcinoma. Cancer Treat Rev 2008, 34(5):407-426. [Pubmed]
-
Meskawi M, Sun M, Trinh QD, Bianchi M, Hansen J, Tian Z, Rink M, Ismail S, Shariat SF, Montorsi F et al: A review of integrated staging systems for renal cell carcinoma. Eur Urol 2012, 62(2):303-314. [Pubmed]
-
Moch H, Artibani W, Delahunt B, Ficarra V, Knuechel R, Montorsi F, Patard JJ, Stief CG, Sulser T, Wild PJ: Reassessing the current UICC/AJCC TNM staging for renal cell carcinoma. Eur Urol 2009, 56(4):636-643. [Pubmed]
-
Paner GP, Stadler WM, Hansel DE, Montironi R, Lin DW, Amin MB: Updates in the Eighth Edition of the Tumor-Node-Metastasis Staging Classification for Urologic Cancers. Eur Urol 2018, 73(4):560-569. [Pubmed]
-
Edge SB BD, Compton CC, et al. : AJCC cancer staging manual. ed. 7. New York, NY: Springer Science and Business Media 2010.
-
59.M H: TNM classification of malignant tumors. 3 rd ed. Geneva, Switzerland: International Union Against Cancer 1978:109-112.
-
Sobin LH WC, editors. : TNM classification of malignant tumours. 5th ed. New York, NY: Wiley-Liss 1997.
-
Hafez KS, Fergany AF, Novick AC: Nephron sparing surgery for localized renal cell carcinoma: impact of tumor size on patient survival, tumor recurrence and TNM staging. J Urol 1999, 162(6):1930-1933. [Pubmed]
-
Ficarra V, Schips L, Guille F, Li G, De La Taille A, Prayer Galetti T, Cindolo L, Novara G, Zigeuner RE, Bratti E et al: Multiinstitutional European validation of the 2002 TNM staging system in conventional and papillary localized renal cell carcinoma. Cancer 2005, 104(5):968-974. [Pubmed]
-
Greene FL PD, Fleming ID, et al. editors. : AJCC cancer staging manual. 6th ed. New York, NY: Springer 2002.
-
Roberts WW, Bhayani SB, Allaf ME, Chan TY, Kavoussi LR, Jarrett TW: Pathological stage does not alter the prognosis for renal lesions determined to be stage T1 by computerized tomography. J Urol 2005, 173(3):713-715. [Pubmed]
-
Jeon HG, Jeong IG, Kwak C, Kim HH, Lee SE, Lee E: Reevaluation of renal cell carcinoma and perirenal fat invasion only. J Urol 2009, 182(5):2137-2143. [Pubmed]
-
Han KR, Bui MH, Pantuck AJ, Freitas DG, Leibovich BC, Dorey FJ, Zisman A, Janzen NK, Mukouyama H, Figlin RA et al: TNM T3a renal cell carcinoma: adrenal gland involvement is not the same as renal fat invasion. J Urol 2003, 169(3):899-903; discussion 903-894. [Pubmed]
-
Suer E, Ergun G, Baltaci S, Beduk Y: Does renal capsular invasion have any prognostic value in localized renal cell carcinoma? J Urol 2008, 180(1):68-71. [Pubmed]
-
Bonsib SM, Gibson D, Mhoon M, Greene GF: Renal sinus involvement in renal cell carcinomas. Am J Surg Pathol 2000, 24(3):451-458. [Pubmed]
-
Thompson RH, Leibovich BC, Cheville JC, Webster WS, Lohse CM, Kwon ED, Frank I, Zincke H, Blute ML: Is renal sinus fat invasion the same as perinephric fat invasion for pT3a renal cell carcinoma? J Urol 2005, 174(4 Pt 1):1218-1221. [Pubmed]
-
Margulis V, Tamboli P, Matin SF, Meisner M, Swanson DA, Wood CG: Location of extrarenal tumor extension does not impact survival of patients with pT3a renal cell carcinoma. J Urol 2007, 178(5):1878-1882. [Pubmed]
-
Jeong IG, Jeong CW, Hong SK, Kwak C, Lee E, Lee SE: Prognostic implication of capsular invasion without perinephric fat infiltration in localized renal cell carcinoma. Urology 2006, 67(4):709-712. [Pubmed]
-
Cho HJ, Kim SJ, Ha US, Hong SH, Kim JC, Choi YJ, Hwang TK: Prognostic value of capsular invasion for localized clear-cell renal cell carcinoma. Eur Urol 2009, 56(6):1006-1012. [Pubmed]
-
Klaver S, Joniau S, Suy R, Oyen R, Van Poppel H: Analysis of renal cell carcinoma with subdiaphragmatic macroscopic venous invasion (T3b). BJU Int 2008, 101(4):444-449. [Pubmed]
-
Martinez-Salamanca JI, Huang WC, Millan I, Bertini R, Bianco FJ, Carballido JA, Ciancio G, Hernandez C, Herranz F, Haferkamp A et al: Prognostic impact of the 2009 UICC/AJCC TNM staging system for renal cell carcinoma with venous extension. Eur Urol 2011, 59(1):120-127. [Pubmed]
-
Kim HL, Zisman A, Han KR, Figlin RA, Belldegrun AS: Prognostic significance of venous thrombus in renal cell carcinoma. Are renal vein and inferior vena cava involvement different? J Urol 2004, 171(2 Pt 1):588-591. [Pubmed]
-
Klatte T, Pantuck AJ, Riggs SB, Kleid MD, Shuch B, Zomorodian N, Kabbinavar FF, Belldegrun AS: Prognostic factors for renal cell carcinoma with tumor thrombus extension. J Urol 2007, 178(4 Pt 1):1189-1195; discussion 1195. [Pubmed]
-
Karakiewicz PI, Trinh QD, Bhojani N, Bensalah K, Salomon L, de la Taille A, Tostain J, Cindolo L, Altieri V, Ficarra V et al: Renal cell carcinoma with nodal metastases in the absence of distant metastatic disease: prognostic indicators of disease-specific survival. Eur Urol 2007, 51(6):1616-1624. [Pubmed]
-
Terrone C, Cracco C, Porpiglia F, Bollito E, Scoffone C, Poggio M, Berruti A, Ragni F, Cossu M, Scarpa RM et al: Reassessing the current TNM lymph node staging for renal cell carcinoma. Eur Urol 2006, 49(2):324-331. [Pubmed]
-
Leibovich BC, Cheville JC, Lohse CM, Zincke H, Frank I, Kwon ED, Merchan JR, Blute ML: A scoring algorithm to predict survival for patients with metastatic clear cell renal cell carcinoma: a stratification tool for prospective clinical trials. J Urol 2005, 174(5):1759-1763; discussion 1763. [Pubmed]
-
Goncalves PD, Srougi M, Dall'lio MF, Leite KR, Ortiz V, Hering F: Low clinical stage renal cell carcinoma: relevance of microvascular tumor invasion as a prognostic parameter. J Urol 2004, 172(2):470-474. [Pubmed]
-
Madbouly K, Al-Qahtani SM, Ghazwani Y, Al-Shaibani S, Mansi MK: Microvascular tumor invasion: prognostic significance in low-stage renal cell carcinoma. Urology 2007, 69(4):670-674. [Pubmed]
-
Delahunt B, Sika-Paotonu D, Bethwaite PB, McCredie MR, Martignoni G, Eble JN, Jordan TW: Fuhrman grading is not appropriate for chromophobe renal cell carcinoma. Am J Surg Pathol 2007, 31(6):957-960. [Pubmed]
-
Sika-Paotonu D, Bethwaite PB, McCredie MR, William Jordan T, Delahunt B: Nucleolar grade but not Fuhrman grade is applicable to papillary renal cell carcinoma. Am J Surg Pathol 2006, 30(9):1091-1096. [Pubmed]
-
Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C et al: Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463(7279):360-363. [Pubmed]
-
Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, Davies H, Jones D, Lin ML, Teague J et al: Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011, 469(7331):539-542. [Pubmed]
-
Kaelin WG: Von Hippel-Lindau disease. Annu Rev Pathol 2007, 2:145-173. [Pubmed]
-
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG, Jr.: HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001, 292(5516):464-468. [Pubmed]
-
Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ et al: Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292(5516):468-472. [Pubmed]
-
Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A et al: C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107(1):43-54. [Pubmed]
-
Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML: Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002, 295(5556):858-861. [Pubmed]
-
Schofield CJ, Ratcliffe PJ: Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 2004, 5(5):343-354. [Pubmed]
-
Nabi S, Kessler ER, Bernard B, Flaig TW, Lam ET: Renal cell carcinoma: a review of biology and pathophysiology. F1000Res 2018, 7:307. [Pubmed]
-
Guo G, Gui Y, Gao S, Tang A, Hu X, Huang Y, Jia W, Li Z, He M, Sun L et al: Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat Genet 2011, 44(1):17-19. [Pubmed]
-
Cancer Genome Atlas Research N: Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499(7456):43-49. [Pubmed]
-
Kapur P, Pe?a-Llopis S, Christie A, Zhrebker L, Pavía-Jiménez A, Rathmell WK, Xie X-J, Brugarolas J: Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: a retrospective analysis with independent validation. The Lancet Oncology 2013, 14(2):159-167. [Pubmed]
-
Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, Shimamura T, Sato-Otsubo A, Nagae G, Suzuki H et al: Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet 2013, 45(8):860-867. [Pubmed]
-
Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR et al: Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 2014, 46(3):225-233. [Pubmed]
-
Nam SJ, Lee C, Park JH, Moon KC: Decreased PBRM1 expression predicts unfavorable prognosis in patients with clear cell renal cell carcinoma. Urol Oncol 2015, 33(8):340 e349-316. [Pubmed]
-
Biegel JA, Busse TM, Weissman BE: SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C Semin Med Genet 2014, 166C(3):350-366. [Pubmed]
-
Hauer MH, Gasser SM: Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev 2017, 31(22):2204-2221. [Pubmed]
-
Conaway RC, Conaway JW: The INO80 chromatin remodeling complex in transcription, replication and repair. Trends Biochem Sci 2009, 34(2):71-77. [Pubmed]
-
de Cubas AA, Rathmell WK: Epigenetic modifiers: activities in renal cell carcinoma. Nat Rev Urol 2018. [Pubmed]
-
Zhuang Z, Park WS, Pack S, Schmidt L, Vortmeyer AO, Pak E, Pham T, Weil RJ, Candidus S, Lubensky IA et al: Trisomy 7-harbouring non-random duplication of the mutant MET allele in hereditary papillary renal carcinomas. Nat Genet 1998, 20(1):66-69. [Pubmed]
-
Fischer J, Palmedo G, von Knobloch R, Bugert P, Prayer-Galetti T, Pagano F, Kovacs G: Duplication and overexpression of the mutant allele of the MET proto-oncogene in multiple hereditary papillary renal cell tumours. Oncogene 1998, 17(6):733-739. [Pubmed]
-
Jones RG, Thompson CB: Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev 2009, 23(5):537-548. [Pubmed]
-
Linehan WM, Srinivasan R, Schmidt LS: The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol 2010, 7(5):277-285. [Pubmed]
-
Crino PB, Nathanson KL, Henske EP: The tuberous sclerosis complex. N Engl J Med 2006, 355(13):1345-1356. [Pubmed]
-
Kwiatkowski DJ, Choueiri TK, Fay AP, Rini BI, Thorner AR, de Velasco G, Tyburczy ME, Hamieh L, Albiges L, Agarwal N et al: Mutations in TSC1, TSC2, and MTOR Are Associated with Response to Rapalogs in Patients with Metastatic Renal Cell Carcinoma. Clin Cancer Res 2016, 22(10):2445-2452. [Pubmed]
-
Laplante M, Sabatini DM: mTOR signaling in growth control and disease. Cell 2012, 149(2):274-293. [Pubmed]
-
Durinck S, Stawiski EW, Pavia-Jimenez A, Modrusan Z, Kapur P, Jaiswal BS, Zhang N, Toffessi-Tcheuyap V, Nguyen TT, Pahuja KB et al: Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet 2015, 47(1):13-21. [Pubmed]
-
Warburg O: On the origin of cancer cells. Science 1956, 123(3191):309-314. [Pubmed]
-
Weiss RH, Lin PY: Kidney cancer: identification of novel targets for therapy. Kidney Int 2006, 69(2):224-232. [Pubmed]
-
Cantor JR, Sabatini DM: Cancer cell metabolism: one hallmark, many faces. Cancer Discov 2012, 2(10):881-898. [Pubmed]
-
Hanahan D, Weinberg RA: Hallmarks of cancer: the next generation. Cell 2011, 144(5):646-674. [Pubmed]
-
Ozcan A, Shen SS, Zhai QJ, Truong LD: Expression of GLUT1 in primary renal tumors: morphologic and biologic implications. Am J Clin Pathol 2007, 128(2):245-254. [Pubmed]
-
Perroud B, Ishimaru T, Borowsky AD, Weiss RH: Grade-dependent proteomics characterization of kidney cancer. Mol Cell Proteomics 2009, 8(5):971-985. [Pubmed]
-
Wettersten HI, Hakimi AA, Morin D, Bianchi C, Johnstone ME, Donohoe DR, Trott JF, Aboud OA, Stirdivant S, Neri B et al: Grade-Dependent Metabolic Reprogramming in Kidney Cancer Revealed by Combined Proteomics and Metabolomics Analysis. Cancer Res 2015, 75(12):2541-2552. [Pubmed]
-
Hakimi AA, Reznik E, Lee CH, Creighton CJ, Brannon AR, Luna A, Aksoy BA, Liu EM, Shen R, Lee W et al: An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 2016, 29(1):104-116. [Pubmed]
-
Catchpole G, Platzer A, Weikert C, Kempkensteffen C, Johannsen M, Krause H, Jung K, Miller K, Willmitzer L, Selbig J et al: Metabolic profiling reveals key metabolic features of renal cell carcinoma. J Cell Mol Med 2011, 15(1):109-118. [Pubmed]
-
Baba M, Furihata M, Hong SB, Tessarollo L, Haines DC, Southon E, Patel V, Igarashi P, Alvord WG, Leighty R et al: Kidney-targeted Birt-Hogg-Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J Natl Cancer Inst 2008, 100(2):140-154. [Pubmed]
-
Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN et al: Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287(5454):848-851. [Pubmed]
-
Dibble CC, Manning BD: Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol 2013, 15(6):555-564. [Pubmed]
-
von Roemeling CA, Marlow LA, Wei JJ, Cooper SJ, Caulfield TR, Wu K, Tan WW, Tun HW, Copland JA: Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin Cancer Res 2013, 19(9):2368-2380. [Pubmed]
-
Yoon CY, Shim YJ, Kim EH, Lee JH, Won NH, Kim JH, Park IS, Yoon DK, Min BH: Renal cell carcinoma does not express argininosuccinate synthetase and is highly sensitive to arginine deprivation via arginine deiminase. Int J Cancer 2007, 120(4):897-905. [Pubmed]
-
Pedoeem A, Azoulay-Alfaguter I, Strazza M, Silverman GJ, Mor A: Programmed death-1 pathway in cancer and autoimmunity. Clin Immunol 2014, 153(1):145-152. [Pubmed]
-
Ribas A, Wolchok JD: Cancer immunotherapy using checkpoint blockade. Science 2018, 359(6382):1350-1355. [Pubmed]
-
Atkins MB, Tannir NM: Current and emerging therapies for first-line treatment of metastatic clear cell renal cell carcinoma. Cancer Treat Rev 2018, 70:127-137. [Pubmed]
-
Hammers HJ, Plimack ER, Infante JR, Rini BI, McDermott DF, Lewis LD, Voss MH, Sharma P, Pal SK, Razak ARA et al: Safety and Efficacy of Nivolumab in Combination With Ipilimumab in Metastatic Renal Cell Carcinoma: The CheckMate 016 Study. J Clin Oncol 2017, 35(34):3851-3858. [Pubmed]
-
Husain FZ, Badani KK, Sfakianos JP, Mehrazin R: Emerging surgical treatments for renal cell carcinoma. Future Oncol 2016, 12(7):921-929. [Pubmed]
-
Ljungberg B, Bensalah K, Canfield S, Dabestani S, Hofmann F, Hora M, Kuczyk MA, Lam T, Marconi L, Merseburger AS et al: EAU guidelines on renal cell carcinoma: 2014 update. Eur Urol 2015, 67(5):913-924. [Pubmed]
-
Campbell SC, Novick AC, Belldegrun A, Blute ML, Chow GK, Derweesh IH, Faraday MM, Kaouk JH, Leveillee RJ, Matin SF et al: Guideline for management of the clinical T1 renal mass. J Urol 2009, 182(4):1271-1279. [Pubmed]
-
Hadjipavlou M, Khan F, Fowler S, Joyce A, Keeley FX, Sriprasad S, Endourology BSo, Oncology: Partial vs radical nephrectomy for T1 renal tumours: an analysis from the British Association of Urological Surgeons Nephrectomy Audit. BJU Int 2016, 117(1):62-71. [Pubmed]
-
Mir MC, Derweesh I, Porpiglia F, Zargar H, Mottrie A, Autorino R: Partial Nephrectomy Versus Radical Nephrectomy for Clinical T1b and T2 Renal Tumors: A Systematic Review and Meta-analysis of Comparative Studies. Eur Urol 2017, 71(4):606-617. [Pubmed]
-
Kunath F, Schmidt S, Krabbe LM, Miernik A, Dahm P, Cleves A, Walther M, Kroeger N: Partial nephrectomy versus radical nephrectomy for clinical localised renal masses. Cochrane Database Syst Rev 2017, 5:CD012045. [Pubmed]
-
Pak JS, Lee JJ, Bilal K, Finkelstein M, Palese MA: Utilization trends and outcomes up to 3 months of open, laparoscopic, and robotic partial nephrectomy. J Robot Surg 2017, 11(2):223-229. [Pubmed]
-
Luciani LG, Chiodini S, Mattevi D, Cai T, Puglisi M, Mantovani W, Malossini G: Robotic-assisted partial nephrectomy provides better operative outcomes as compared to the laparoscopic and open approaches: results from a prospective cohort study. J Robot Surg 2017, 11(3):333-339. [Pubmed]
-
Ng AM, Shah PH, Kavoussi LR: Laparoscopic Partial Nephrectomy: A Narrative Review and Comparison with Open and Robotic Partial Nephrectomy. J Endourol 2017, 31(10):976-984. [Pubmed]
-
Leow JJ, Heah NH, Chang SL, Chong YL, Png KS: Outcomes of Robotic versus Laparoscopic Partial Nephrectomy: an Updated Meta-Analysis of 4,919 Patients. J Urol 2016, 196(5):1371-1377. [Pubmed]
-
Bragayrac LA, Abbotoy D, Attwood K, Darwiche F, Hoffmeyer J, Kauffman EC, Schwaab T: Outcomes of Minimal Invasive vs Open Radical Nephrectomy for the Treatment of Locally Advanced Renal-Cell Carcinoma. J Endourol 2016, 30(8):871-876. [Pubmed]
-
Yang DY, Monn MF, Bahler CD, Sundaram CP: Does robotic assistance confer an economic benefit during laparoscopic radical nephrectomy? J Urol 2014, 192(3):671-676. [Pubmed]
-
McDermott DF, Regan MM, Clark JI, Flaherty LE, Weiss GR, Logan TF, Kirkwood JM, Gordon MS, Sosman JA, Ernstoff MS et al: Randomized phase III trial of high-dose interleukin-2 versus subcutaneous interleukin-2 and interferon in patients with metastatic renal cell carcinoma. J Clin Oncol 2005, 23(1):133-141. [Pubmed]
-
Galligioni E, Francini M, Quaia M, Carbone A, Spada A, Sacco C, Favaro D, Santarosa M, Carmignani G, Di Donna D et al: Randomized study of adjuvant immunotherapy with autologous tumor cells and BCG in renal cancer. Ann N Y Acad Sci 1993, 690:367-369. [Pubmed]
-
Repmann R, Goldschmidt AJ, Richter A: Adjuvant therapy of renal cell carcinoma patients with an autologous tumor cell lysate vaccine: a 5-year follow-up analysis. Anticancer Res 2003, 23(2A):969-974. [Pubmed]
-
Escudier B, Sharma P, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER et al: CheckMate 025 Randomized Phase 3 Study: Outcomes by Key Baseline Factors and Prior Therapy for Nivolumab Versus Everolimus in Advanced Renal Cell Carcinoma. Eur Urol 2017, 72(6):962-971. [Pubmed]
-
Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER et al: Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 2015, 373(19):1803-1813. [Pubmed]
-
Atkins MB, Plimack ER, Puzanov I, Fishman MN, McDermott DF, Cho DC, Vaishampayan U, George S, Olencki TE, Tarazi JC et al: Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: a non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial. The Lancet Oncology 2018, 19(3):405-415. [Pubmed]
-
Alsharedi M, Katz H: Check point inhibitors a new era in renal cell carcinoma treatment. Med Oncol 2018, 35(6):85. [Pubmed]
-
Ledford H: Melanoma drug wins US approval. Nature 2011, 471(7340):561. [Pubmed]
-
Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, Plimack ER, Barthelemy P, Porta C, George S et al: Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med 2018, 378(14):1277-1290. [Pubmed]
-
Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST et al: Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 2007, 356(2):115-124. [Pubmed]
-
Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA et al: Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007, 356(2):125-134. [Pubmed]
-
Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C, Chevreau C, Filipek M, Melichar B, Bajetta E et al: Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet 2007, 370(9605):2103-2111. [Pubmed]
-
Powles T, Albiges L, Staehler M, Bensalah K, Dabestani S, Giles RH, Hofmann F, Hora M, Kuczyk MA, Lam TB et al: Updated European Association of Urology Guidelines: Recommendations for the Treatment of First-line Metastatic Clear Cell Renal Cancer. European Urology 2018, 73(3):311-315. [Pubmed]
-
Calvo E, Porta C, Grunwald V, Escudier B: The Current and Evolving Landscape of First-Line Treatments for Advanced Renal Cell Carcinoma. Oncologist 2018. [Pubmed]
-
Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, Heng DY, Larkin J, Ficarra V: Renal cell carcinoma. Nat Rev Dis Primers 2017, 3:17009. [Pubmed]
-
Fernandez-Pello S, Hofmann F, Tahbaz R, Marconi L, Lam TB, Albiges L, Bensalah K, Canfield SE, Dabestani S, Giles RH et al: A Systematic Review and Meta-analysis Comparing the Effectiveness and Adverse Effects of Different Systemic Treatments for Non-clear Cell Renal Cell Carcinoma. Eur Urol 2017, 71(3):426-436. [Pubmed]
Cite this article as: Zhang P, Ro JY: Renal cell carcinoma. Ann Urol Oncol 2018, 1(1): 1-18. doi: 10.32948/auo.2018.11.1
Annals of urologic oncology
p-ISSN: 2617-7765, e-ISSN: 2617-7773
 Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.