
 Submit Manuscript
Submit ManuscriptMutations in Cancer Driver Genes: An Insight into Prostate Cancer Progression
Sahdeo Prasad1, Sanjay K Srivastava1
1Department of Immunotherapeutics and Biotechnology, Texas Tech University Health Sciences Center, Abilene, TX 79601, USA.
Correspondence: Sanjay K. Srivastava or Sahdeo Prasad (Department of Immunotherapeutics and Biotechnology, Texas Tech University Health Sciences Center, Suite 1305, 1718 Pine Street, Abilene, Texas 79601, USA; Email: sanjay.srivastava@ttuhsc.edu (SKS), sahdeo.prasad@ttuhsc.edu (SP).)
Annals of Urologic Oncology 2019, 2(2): 89-95. https://doi.org/10.32948/auo.2019.09.12
Received: 24 Jun 2019 | Accepted: 12 Sep 2019 | Published online: 22 Sep 2019 
Prostate cancer is one of the most common uro-oncological disease in men and is globally leading cause of cancer related deaths in males. The somatic mutation has a strong link in the occurrence of cancer. Mutation in the oncogenes and tumor suppressor genes that alter key cellular functions can lead to prostate cancer initiation and progression. Whole genome sequencing has identified numerous genetic alternations and further provided a detail view of the mutations in genes that drive progression of prostate cancer. TP53, SPOP, PTEN, ATM, AR, CTNNB1, FOXA1, KMT2D, BRACA2 and APC were found as frequently mutated genes in prostate cancer. Using data from cBioPortal and PubMed, this review summarizes the status and possible impact of mutations in these driver genes on survival, progression, and metastasis of prostate cancer. This study will contribute a better understanding of biological basis for clinical variability in prostate cancer patients and may provide new genetic diagnostic markers and drug targets.
Key words mutation, prostate cancer, driver genes, diagnostic and therapeutic targets
Although analysis of the prostate cancer genome began at the early of this century, recently the tremendous effort on whole genome sequencing represented a breakthrough in the analysis of hundreds of tumors [2, 3]. cBioPortal for Cancer Genomics, a Web-based resource of genomic data, provides visualization, analysis and share large-scale cancer genomic data sets from thousands of patients worldwide. In this review using the cBio Cancer Genomics Portal (http://cbioportal.org) data, we will discuss the highly mutated driver genes of prostate tumorigenesis. Somatic mutations in the genes that drive tumorigenesis are called driver genes. Although as of now cBio Cancer Genomics Portal comprises data of 18 studies on prostate adenocarcinoma, we will analyze data from most recent studies that include Prostate Adenocarcinoma (MSKCC/DFCI 2018), The Metastatic Prostate Cancer Project (Provisional 2018), and Metastatic Prostate Adenocarcinoma (SU2C/PCF Dream Team 2019). In 2018, MSKCC/DFCI performed sequencing on 1013 (680 primary and 333 metastatic) cases of prostate adenocarcinoma; while Metastatic Prostate Cancer Project analyzed genome sequencing of 19 samples in the same year. Recently SU2C/PCF Dream Team has sequenced 444 samples and provided a genetic landscape of prostate adenocarcinoma for understanding this complex disease. The data of these studies demonstrated that prostate adenocarcinoma has several genomic alterations, which are responsible for tumorigenesis. This article will focus on highly mutated driver genes (≥3%) in prostate adenocarcinoma. Table 1 summarizes the highly mutated genes in these three recent study datasets. The findings of these studies demonstrated that the most frequently and highly mutated genes in prostate adenocarcinoma are TP53, SPOP, FOXA1, ATM, AR, PTEN, CTNNB1, KMT2D, BRACA2, and APC. However, certain cancer drivers are mutated in a small fraction of prostate tumors [4].
Although prostate adenocarcinoma has lesser mutations than other cancer types, they have a high rate of genomic instability and chromosomal rearrangements [5]. However, several studies suggest that compared to primary tumors, metastatic CRPC have about five times more mutations [6-8]. As per the cBio Cancer Genomics Portal database, the most frequent genomic mutation in prostate adenocarcinoma is TP53 (18-42%). Besides mutation, TP53 has other genomic alterations such as deletion, amplification etc. that contribute 2-10% of instability. The role of TP53 and other mutated driver genes in tumorigenesis of the prostate will be discussed.
To discover the genetic driver of prostate cancer, large-scale genome sequencing has been done and identified a number of mutated genes. We will discuss some important frequently mutated genes, which are mutated in approximately 3% of prostate tumor samples and that drive prostate cancer progression.
TP53
The TP53 tumor suppressor gene is one of the most frequently mutated genes in human malignancies [9]. According to the PRAD (MSKCC/DFCI 2018) study as mentioned in the cBioportal dataset, 20.53% of prostate tumors had altered genomes. Moreover, 18.56% of prostate tumors (188 out of 1013 samples) were with mutated TP53. However, The MPC Project study revealed that 52.63% of prostate tumors have various alterations including a mutation in TP53 gene; while 42.11% of tumors have only mutations (8 samples out of 19) in their genome. Recently the Prostate (SUTC 2019) study revealed that 36.26% of prostate tumors (161 out of 444 samples) have a mutation in their TP53 gene whereas 40.09% of tumors have various alterations in their genome. Besides these, another study has shown that 35.6% (32 out of 90 patients) tumor samples of prostatectomy contain one or more TP53 mutations. However, among them, 40.6% of prostate tumors with TP53 mutations were associated with cancer progression [10].
The p53 protein, sequence-specific transcription factor, plays a key role in maintaining genomic stability and preventing tumorigenesis by arresting the cell cycle, DNA repair, and apoptosis under stress condition [11]. Mutation in the TP53 gene is known to imbalance the cell homeostasis that drives toward tumorigenesis. It has been shown that a single mutation in TP53 (R270H) was sufficient to induce prostate cancer in mice [12]. Ecke et al. [10] reported that 40.6% of mutated TP53 leads to tumor progression in prostate cancer patients. Besides these, mutation or deletion of TP53 also increases the risk of prostate cancer metastasis andrecurrence and recurrence [13, 14]. Thus, the mutation in the TP53 gene drive prostate cancer initiation, progression, metastasis, and recurrence.
SPOP
Prostate tumorigenesis is facilitated by frequent mutation in SPOP (Speckle-type POZ Protein) gene as elucidated by genomic sequencing analysis [3]. As mentioned in Table 1, the PRAD study revealed that 9.38% (95 samples out of 1013) prostate tumors have alterations in the SPOP gene. These 9.38% alterations represent the only mutation in the SPOP gene. Genomic sequencing effort by MPC project also revealed that 26.32% (5 samples out of 19) tumors have alteration in the SPOP gene. Moreover, the mutant SPOP gene was in 15.79% prostate tumor samples. Further Prostate (SUTC 2019) study as mentioned in cBioportal database showed that 10.59% (47 samples out of 444) tumors contain various alterations in the SPOP gene whereas mutant SPOP gene was in 5.41% (24 cases) tumors. These databases suggest the importance of SPOP gene mutation in prostate tumorigenesis.
SPOP is an adapter component of the CUL3 E3-ligase complex, which degrades its target by adding an ubiquitin tag onto them.The known substrates of SPOP are androgen receptor [15], the steroid co-activator SRC3 [16], and the DEK and ERG oncogenes [17, 18]. It acts as tumor suppressive. As stated above SPOP has up to 15.79% mutation in primary prostate tumors [3], suggesting that SPOP may act as a potential driver of prostate cancer. Mutated SPOP are unable to bind and promote the degradation of substrates, which leads to increased prostate cancer cell proliferation, invasion, and resistance to antiandrogen therapy of prostate cancer [3]. In a mouse model, mutations in SPOP result in prostate neoplasia through the activation of PI3K/mTOR signaling and upregulation of its substrate SRC3 [19].
PTEN
Phosphatase and tensin homologue (PTEN), a tumor suppressor, is one of the most frequently mutated gene in human cancer [20]. About 40-50% of mutation have shown to occur in primary prostate tumors [21, 22]. A recent report of PRAD study recorded in cBioportal database states that 4.24% (43 samples out of 1013) of prostate tumors have mutated PTEN, however 16.49% of prostate tumor samples include other alteration in PTEN gene. The MPC project also reported that 5.26% of samples had a mutated PTEN gene whereas 15.79% (3 samples out of 19) samples contain multiple gene alterations. Genome sequencing analysis performed by SU2C 2019 revealed that 6.31% of prostate tumors comprise a PTEN gene mutation. However, 32.66% (145 samples out of 444) of tumors contain various types of alterations in the PTEN genome. These data indicate that mutation in the PTEN gene is one of the main reason of prostate tumorigenesis.
PTEN deactivates PI3K/Akt signaling pathway by dephosphorylating lipid signaling molecules. However, the loss of PTEN results in growth, survival and poor prognosis of prostate cancer [23]. Besides these, accumulated evidence suggests that the deletion of PTEN is linked with metastasis of prostate cancer and higher Gleason grade of the disease [24, 25]. Loss of PTEN also results in the recurrence of the disease after therapy and may lead to the death of prostate cancer patients [26, 27]. Moreover, PIK3CA mutation and PTEN loss coexist in prostate cancer that cooperates and accelerates tumorigenesis and further facilitate castrate-resistant prostate cancers [28]. This acts synergistically and thus it could be therapeutic/prognostic approach against prostate cancer, particularly CRPC.
ATM
Ataxia-telangiectasia mutated (ATM) is a DNA repair gene, germline mutations of which responsible for a variety of human hereditary diseases and predisposition to cancer [29]. Genome sequence analysis by PRAD study as mentioned in cBioportal database shows that 4.54% of the 1013 prostate tumor samples have deleterious germline or somatic aberrations. However, 3.75% (38 samples out of 1013) acquire mutated ATM gene. Later gene sequence analysis by SU2C study revealed that 7.88% of 444 tumor samples have alterations in the ATM gene. The mutated ATM gene was found only in 26 cases out of 444 (5.86%) prostate tumor samples. Although the frequency of ATM gene mutation is low, it may play an important role in prostate tumorigenesis.
Mutations in DNA repair genes such as ATM are associated with aggressive prostate cancer. An advanced tumor stage and higher Gleason grade group is also found in prostate cancer patients with an enriched ATM mutation gene [30]. ATM pathway directly participates in the ionizing radiation-induced cell proliferation inhibition. A study has shown that the inactivation of the ATM pathway by using ATM inhibitors abolishes ionizing radiation-induced cell proliferation inhibition in prostate cancer cell lines [31]. Besides these, the frequency of aberrations in ATM is higher in CRPC [7], which indicates ATM mutation has a potential role in mediating castration resistance.
CTNNB1
CTNNB1 gene encodes the cytoplasmic protein β-Catenin, which is known to be crucial in many steps of prostate tumorigenesis. It has been previously reported that 5% of prostate tumors harbor CTNNB1 mutations [32]. The PRAD (MSKCC/DFCI) study as mentioned in cBioportal database reported that 3.95% of 1013 prostate tumor samples contain alterations in their CTNNB1 genome. However, only 2.96% (30 samples out of 1013) have mutated CTNNB1. Further, the SU2C study performed genome sequence analysis of 444 prostate tumor samples and revealed that 8.56% of tumors contain alteration in the CTNNB1 genome. However, only 4.28% (19 samples out of 444) have mutated CTNNB1 gene. A genome sequence analysis done in 2019 (MPC project) has also reported that 10.53% (2 samples out of 19) of prostate tumors have CTNNB1 gene alterations that include mutation and amplification. Among them, 1 sample (5.26%) contains a mutation in the CTNNB1 gene.
The CTNNB1 encoded β-Catenin protein participates in the formation of the adherens junction with E-Cadherin and is also an important component of canonical WNT signaling [33]. It has been shown that CTNNB1 gene mutation in prostate cancer activates the WNT/β-catenin signaling cascade [34]. In a study, it was observed that the conditional deletion of the CTNNB1 gene in the prostates of experimental mice during embryonic stages resulted in decreased prostatic budding and also abolished development of the prostate [35]. Thus, β-Catenin is essential for prostate development, however its overexpression can promote invasive prostate cancer [36].
AR
The androgen receptor (AR) is a steroid receptor, characterized as a ligand-dependent transcription factor. AR gene mutation in the primary tumor is usually rare but it is higher in advanced and androgen-independent tumors. In a study, out of 99 early-stage prostate tumors no mutation was observed in the AR gene. However, 21% of advanced-stage prostate tumors found to have a mutation in the AR gene [37]. Similarly, the PRAD study reported only 3.95% (40 samples out of 1013) of prostate tumors have a mutation in the AR gene while total 16.49% of samples found to have multiple types of alteration in AR genome. SU2C also reported 44 samples (9.91%) out of 444 prostate tumors with mutation and 58.78% tumors have multiple types of genomic alterations including a mutation in the AR gene. However, the MPC project found no prostate tumor sample with AR mutation among the 19 analyzed samples but 2 samples had amplification in the AR gene.
AR signaling remains the ultimate driver of prostate tumorigenesis. The mutation or amplification of the AR gene further helps in the constitutive expression of AR protein that promotes sustained activation of the AR signaling pathway. In support, Shi et al. also found gain of function in 20 out of 44 such mutations in AR gene [38]. AR amplification, AR mutations, or constitutive activation of AR signaling also causes resistance of prostate cancer to therapeutic drugs, which targets AR signaling [1, 39]. Even in the androgen-depleted environment, prostate tumor cells with such mutations or amplifications are likely to provide a growth advantage [40].
FOXA1
Forkhead box A1 (FOXA1) is a transcription factor involve in growth and differentiation of prostate cancer. Unlike AR, mutations in FOXA1 have been found in both primary tumors as well as metastatic prostate tumors. Earlier whole genome sequencing analysis showed that FOXA1 mutations occur in 3.4 – 5.2% of prostate tumors [3, 21]. However later study by PRAD reported mutation in 6.22% (63 samples out of 1013) of prostate tumors, although 9.38% of tumors contain multiple alterations in the FOXA1 gene. SU2C study as recorded in cBioportal database, revealed that the mutated FOXA1 gene found in 9.01% (40 cases out of 444) tumors while 13.96% samples contain multiple alterations including mutation. Recently the MPC project reported a 5.26% prostate tumor with a mutation in the FOXA1 gene. These latest studies indicate that FOXA1 gene is mutated in more than 5% of prostate tumors, which is much more than previous studies [3, 21].
FOXA1 drives prostate tumorigenesis by interacting with AR. It has been found that prostate tumors with FOXA1 mutation have the highest AR transcriptional activity. In CRPC, AR preferentially binds to FOXA1 mutants [41]. Studies showed that metastatic and high-grade prostate tumors contain amplification of the FOXA1 gene, which indicates alterations in the FOXA1 contribute to metastasis and advancement in disease progression [21, 42, 43]. Mutation in FOXA1 gene results in a gain of function activity [44]. It has been also speculated that mutation in FOXA1 stabilizes the protein that leads to an increase in its DNA binding affinity, resulting in the expression of FOXA1 target proteins [45]. Thus, FOXA1 has a role in prostate tumorigenesis either in the form of wild type or in mutant form.
KMT2D
Histone lysine methyltransferase 2D (KMT2D/MLL2) is an important epigenetic modifier. It catalyzes the methylation of H3K4 [46] and plays an important role in prostate tumorigenesis. Approximately 0-12% of prostate tumors have been found to contain the KMT2D gene with mutation [47]. Supporting this data, the PRAD study also revealed that 6.42% of prostate tumors in 1013 samples have mutated KMT2D gene. The SU2C study also found 8.33% (37 samples out of 444) of prostate tumors with mutations in KMT2D gene. Besides these, the MPC project revealed by whole genome sequence analysis that 10.53% (2 samples out of 19) are with mutated KMT2D gene. This project also found various other types of alterations including a mutation in 21.05% of prostate tumors in the KMT2D gene. However, in Chinese patients, the KMT2D gene was found to be highly mutated (63.04%, 29/46) as analyzed by using disease-targeted sequencing [48].
The expression of KMT2D is essential for tumor cell survival, proliferation, and resistance of various cancers [49, 50]. In prostate cancer, it has been shown that mutation in the KMT2D gene is ‘prostate cancer-specific’ and might involve in the malignant progression from high grade prostatic intraepithelial neoplasia to cancer [51]. Inactivation of the KMT2D gene causes genomic instability in prostate cancer. Also, KMT2D mutated cells display signs of substantial transcription stress that elevate levels of γH2AX, and further frequent mutation [52].
BRACA2
Mutations in the tumor suppressor gene BRCA2 have reported to be a genetic risk factor for prostate cancer. Kote-Jarai et al [53] have shown that germline BRCA2 mutations are present in 1.2 - 2% of prostate cancer cases. However, cBioportal data from recent studies revealed that the BRCA2 gene is mutated in 2.76 - 26.32% of prostate tumor cases. Observation of PRAD study showed that 2.76% (28 cases out of 1013) of prostate tumor have mutated BRCA2 gene and total alterations in BRCA2 gene were in 5.23% of prostate cancer samples. However, SU2C study revealed that 8.33% (37 samples out of 444) of prostate tumors contain mutations in BRCA2 gene and total alterations including mutation were in 12.16% of tumors. Besides these studies, MPC project found 26.32% (5 samples out of 19) of prostate tumor with BRCA2 gene mutation, which is markedly higher than the findings of PRAD and SU2C studies. This difference could be linked with the number of cases used for the analysis.
BRCA2 mutation in prostate cancer patients causes poor prognosis and worse therapeutic clinical outcomes. In addition, BRCA2-mutant tumors can tolerate castration that helps in enabling the tumor to evade androgen deprivation therapy [54]. Germline mutations in the BRCA2 also increase the risk of prostate cancer development. It has been shown that BRCA2 mutation in prostate cancer causes genomic and epigenomic dysregulation of the MED12L/MED12 axis, which is one of the factor of metastatic CRPC [55]. Mutation in the BRCA2 gene also causes a decrease in the overall survival of prostate cancer patients. In a cohort study, Edwards et al. [56] showed that patients with BRCA2 mutation had a median survival of 4.8 years compared to 8.5 years of non-mutated patients. These studies conclude that germline mutations in BRCA2 result in poor prognosis, increases risk, decreases overall survival and various other complications in prostate cancer patients.
APC
APC (Adenomatous Polyposis Coli) is a tumor-suppressor gene. Although its mutation is primarily involved in the early stage of tumor development in the digestive tract, a mutation in the primary and advanced stage of prostate tumors has also been reported [57]. Mutations in APC gene were found to be from 3–10% of prostate cancer samples as observed by various whole genome sequencing studies [58]. A later analysis by PRAD study as reported in cBioportal database showed that 3.26% (33 cases out of 1013) contain mutations in the APC gene while other genomic alterations including mutation were 5.03% in prostate tumors specimens. Further SU2C study revealed that APC genes are mutated in 6.98% (31 cases out of 444) of prostate tumors. This study, however, found 8.11% of prostate tumors with multiple alterations in the APC gene.
The APC acts as an antagonist of the Wnt/β-catenin signaling pathway. Deletion of APC gene causes induction of prostate tumor through elevation of Wnt/β-catenin proteins. The mutation of APC gene also involve in metastasis of prostate cancer [59, 60]. In addition, the deletion of the APC gene causes high-grade prostatic intraepithelial neoplasia (HGPIN) in mouse prostate epithelium, with high penetrance but this does not lead to metastasis [59]. Besides these, methylation of the APC promoter contributes to the activation of Wnt/β-catenin signaling pathway and results in the poor prognosis of prostate cancer [61]. APC has been reported to undergo loss of heterozygosity in around 20% of primary cancers and 90% APC promoters are found to be CpG methylated in prostate tumors [62, 63].
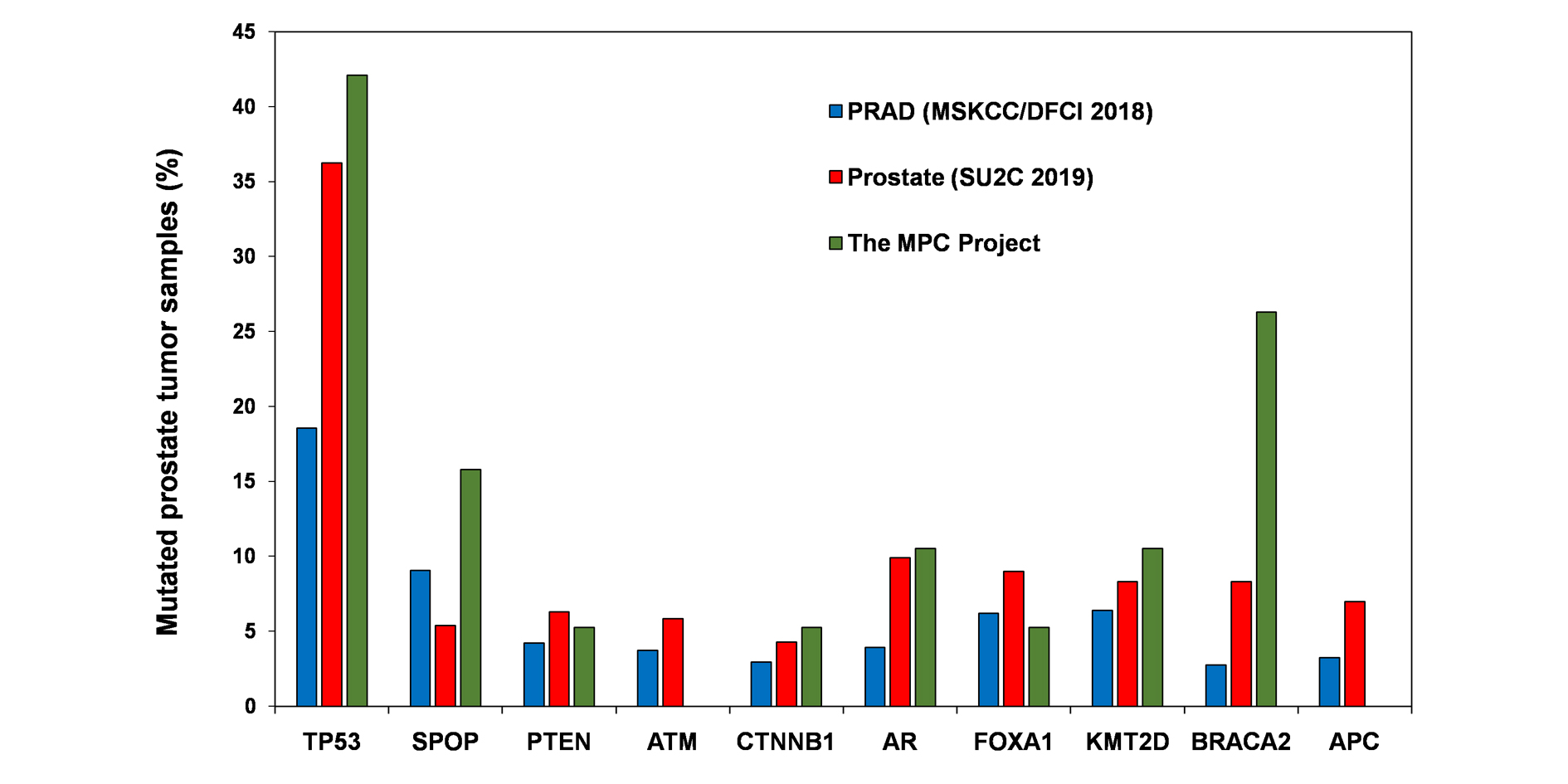 Figure 1: Prostate tumor samples (%) with mutation analyzed by different study groups. Bar diagram is plotted from the data collectively taken from cBioportal dataset.
Figure 1: Prostate tumor samples (%) with mutation analyzed by different study groups. Bar diagram is plotted from the data collectively taken from cBioportal dataset.
|
Table 1: Percent mutated samples analyzed by genome sequencing. |
|||
|
Gene symbol |
PRAD(MSKCC/DFCI 2018) (N = 1013) |
Prostate (SU2C 2019) (N = 444) |
The MPC Project (N = 19) |
|
TP53 |
18.56 |
36.26 |
42.11 |
|
SPOP |
9.08 |
5.41 |
15.79 |
|
PTEN |
4.24 |
6.31 |
5.26 |
|
ATM |
3.75 |
5.86 |
NF |
|
CTNNB1 |
2.96 |
4.28 |
5.26 |
|
AR |
3.95 |
9.91 |
10.53 |
|
FOXA1 |
6.22 |
9.01 |
5.26 |
|
KMT2D |
6.42 |
8.33 |
10.53 |
|
BRACA2 |
2.76 |
8.33 |
26.32 |
|
APC |
3.26 |
6.98 |
NF |
|
Abbreviations: NF-not found |
|||
Considering the status of mutations in the genes that drive carcinogenesis, prostate cancer can be potentially targeted at somatic as well as at the germline level. There would be a possibility to integrate Next-Gen Sequencing to the clinical practice. This approach may provide additional predictive value to the conventional therapy and it may be useful to the prostate cancer patients with distinct clinical manifestation of disease and different response to therapy. Besides these, ultra-deep sequencing of prostate tumors can be also performed to detect rare mutations, which will provide additional information on prostate tumor heterogeneity. Advancement in tumor gene mutation profiling at a personalized level in a clinical setting can also be feasible to translate genome sequencing from bench to bedside, which will advantageous in prostate cancer treatment.
Funding
None.
Ethics approval and consent to participate
This study did not require prior ethics approval or consent from human participants.
Author contributions
SP designed, conceptualized, assembled articles, wrote and revised the manuscript. SKS guided and reviewed the manuscript.
Competing interests
The authors declare no conflict of interest with the work.
-
Coutinho I, Day TK, Tilley WD, Selth LA: Androgen receptor signaling in castration-resistant prostate cancer: a lesson in persistence. Endocr Relat Cancer 2016, 23(12): T179-T197.
-
Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M et al: Punctuated evolution of prostate cancer genomes. Cell 2013, 153(3): 666-677.
-
Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N et al: Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 2012, 44(6): 685-689.
-
Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J et al: The genomic landscapes of human breast and colorectal cancers. Science 2007, 318(5853): 1108-1113.
-
Barbieri CE, Rubin MA: Genomic rearrangements in prostate cancer. Curr Opin Urol 2015, 25(1): 71-76.
-
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G: Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505(7484): 495-501.
-
Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G et al: Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 162(2): 454.
-
Wedge DC, Gundem G, Mitchell T, Woodcock DJ, Martincorena I, Ghori M, Zamora J, Butler A, Whitaker H, Kote-Jarai Z et al: Sequencing of prostate cancers identifies new cancer genes, routes of progression and drug targets. Nat Genet 2018, 50(5): 682-692.
-
Hainaut P, Hernandez T, Robinson A, Rodriguez-Tome P, Flores T, Hollstein M, Harris CC, Montesano R: IARC Database of p53 gene mutations in human tumors and cell lines: updated compilation, revised formats and new visualisation tools. Nucleic Acids Res 1998, 26(1): 205-213.
-
Ecke TH, Schlechte HH, Schiemenz K, Sachs MD, Lenk SV, Rudolph BD, Loening SA: TP53 gene mutations in prostate cancer progression. Anticancer Res 2010, 30(5): 1579-1586.
-
Liu J, Zhang C, Wang XL, Ly P, Belyi V, Xu-Monette ZY, Young KH, Hu W, Feng Z: E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ 2014, 21(11): 1792-1804.
-
Vinall RL, Chen JQ, Hubbard NE, Sulaimon SS, Shen MM, Devere White RW, Borowsky AD: Initiation of prostate cancer in mice by Tp53R270H: evidence for an alternative molecular progression. Dis Model Mech 2012, 5(6): 914-920.
-
Hong MK, Macintyre G, Wedge DC, Van Loo P, Patel K, Lunke S, Alexandrov LB, Sloggett C, Cmero M, Marass F et al: Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat Commun 2015, 6: 6605.
-
Kluth M, Harasimowicz S, Burkhardt L, Grupp K, Krohn A, Prien K, Gjoni J, Hass T, Galal R, Graefen M et al: Clinical significance of different types of p53 gene alteration in surgically treated prostate cancer. Int J Cancer 2014, 135(6): 1369-1380.
-
An J, Wang C, Deng Y, Yu L, Huang H: Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep 2014, 6(4): 657-669.
-
Geng C, He B, Xu L, Barbieri CE, Eedunuri VK, Chew SA, Zimmermann M, Bond R, Shou J, Li C et al: Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc Natl Acad Sci U S A 2013, 110(17): 6997-7002.
-
An J, Ren S, Murphy SJ, Dalangood S, Chang C, Pang X, Cui Y, Wang L, Pan Y, Zhang X et al: Truncated ERG Oncoproteins from TMPRSS2-ERG Fusions Are Resistant to SPOP-Mediated Proteasome Degradation. Mol Cell 2015, 59(6): 904-916.
-
Theurillat JP, Udeshi ND, Errington WJ, Svinkina T, Baca SC, Pop M, Wild PJ, Blattner M, Groner AC, Rubin MA et al: Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science 2014, 346(6205): 85-89.
-
Blattner M, Liu D, Robinson BD, Huang D, Poliakov A, Gao D, Nataraj S, Deonarine LD, Augello MA, Sailer V et al: SPOP Mutation Drives Prostate Tumorigenesis In Vivo through Coordinate Regulation of PI3K/mTOR and AR Signaling. Cancer Cell 2017, 31(3): 436-451.
-
Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, Jen J, Isaacs WB, Bova GS, Sidransky D: Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res 1997, 57(22): 4997-5000.
-
Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC et al: The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487(7406): 239-243.
-
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B et al: Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18(1): 11-22.
-
Chalhoub N, Baker SJ: PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol 2009, 4: 127-150.
-
McMenamin ME, Soung P, Perera S, Kaplan I, Loda M, Sellers WR: Loss of PTEN expression in paraffin-embedded primary prostate cancer correlates with high Gleason score and advanced stage. Cancer Res 1999, 59(17): 4291-4296.
-
Attard G, Swennenhuis JF, Olmos D, Reid AH, Vickers E, A'Hern R, Levink R, Coumans F, Moreira J, Riisnaes R et al: Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res 2009, 69(7): 2912-2918.
-
Choucair K, Ejdelman J, Brimo F, Aprikian A, Chevalier S, Lapointe J: PTEN genomic deletion predicts prostate cancer recurrence and is associated with low AR expression and transcriptional activity. BMC Cancer 2012, 12: 543.
-
Reid AH, Attard G, Ambroisine L, Fisher G, Kovacs G, Brewer D, Clark J, Flohr P, Edwards S, Berney DM et al: Molecular characterisation of ERG, ETV1 and PTEN gene loci identifies patients at low and high risk of death from prostate cancer. Br J Cancer 2010, 102(4): 678-684.
-
Pearson HB, Li J, Meniel VS, Fennell CM, Waring P, Montgomery KG, Rebello RJ, Macpherson AA, Koushyar S, Furic L et al: Identification of Pik3ca Mutation as a Genetic Driver of Prostate Cancer That Cooperates with Pten Loss to Accelerate Progression and Castration-Resistant Growth. Cancer Discov 2018, 8(6): 764-779.
-
Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, Garofalo A, Gulati R, Carreira S, Eeles R et al: Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N Engl J Med 2016, 375(5): 443-453.
-
Marshall CH, Fu W, Wang H, Baras AS, Lotan TL, Antonarakis ES: Prevalence of DNA repair gene mutations in localized prostate cancer according to clinical and pathologic features: association of Gleason score and tumor stage. Prostate Cancer Prostatic Dis 2019, 22(1): 59-65.
-
Li SJ, Liang XY, Li HJ, Yang GZ, Li W, Li Z, Zhou L, Wen X, Yu DH, Cui JW: Low-dose irradiation inhibits proliferation of the p53null type human prostate cancer cells through the ATM/p21 pathway. Int J Mol Med 2018, 41(1): 548-554.
-
Voeller HJ, Truica CI, Gelmann EP: beta-catenin mutations in human prostate cancer. Cancer Research 1998, 58(12): 2520-2523.
-
Pramanik KC, Fofaria NM, Gupta P, Ranjan A, Kim SH, Srivastava SK: Inhibition of beta-catenin signaling suppresses pancreatic tumor growth by disrupting nuclear beta-catenin/TCF-1 complex: critical role of STAT-3. Oncotarget 2015, 6(13): 11561-11574.
-
Katoh M, Katoh M: Molecular genetics and targeted therapy of WNT-related human diseases (Review). Int J Mol Med 2017, 40(3): 587-606.
-
Simons BW, Hurley PJ, Huang Z, Ross AE, Miller R, Marchionni L, Berman DM, Schaeffer EM: Wnt signaling though beta-catenin is required for prostate lineage specification. Dev Biol 2012, 371(2): 246-255.
-
Francis JC, Thomsen MK, Taketo MM, Swain A: beta-catenin is required for prostate development and cooperates with Pten loss to drive invasive carcinoma. PLoS Genet 2013, 9(1): e1003180.
-
Marcelli M, Ittmann M, Mariani S, Sutherland R, Nigam R, Murthy L, Zhao Y, DiConcini D, Puxeddu E, Esen A et al: Androgen receptor mutations in prostate cancer. Cancer Res 2000, 60(4): 944-949.
-
Shi XB, Ma AH, Xia L, Kung HJ, de Vere White RW: Functional analysis of 44 mutant androgen receptors from human prostate cancer. Cancer Res 2002, 62(5): 1496-1502.
-
Mahajan K, Malla P, Lawrence HR, Chen Z, Kumar-Sinha C, Malik R, Shukla S, Kim J, Coppola D, Lawrence NJ et al: ACK1/TNK2 Regulates Histone H4 Tyr88-phosphorylation and AR Gene Expression in Castration-Resistant Prostate Cancer. Cancer Cell 2017, 31(6): 790-803 e798.
-
Feldman BJ, Feldman D: The development of androgen-independent prostate cancer. Nat Rev Cancer 2001, 1(1): 34-45.
-
Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA et al: Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 2011, 474(7351): 390-394.
-
Robbins CM, Tembe WA, Baker A, Sinari S, Moses TY, Beckstrom-Sternberg S, Beckstrom-Sternberg J, Barrett M, Long J, Chinnaiyan A et al: Copy number and targeted mutational analysis reveals novel somatic events in metastatic prostate tumors. Genome Res 2011, 21(1): 47-55.
-
Gerhardt J, Montani M, Wild P, Beer M, Huber F, Hermanns T, Muntener M, Kristiansen G: FOXA1 promotes tumor progression in prostate cancer and represents a novel hallmark of castration-resistant prostate cancer. Am J Pathol 2012, 180(2): 848-861.
-
Adams EJ, Hoover E, Karthaus WR, Sawyers CL: FOXA1 promotes a luminal growth program in prostate cancer [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2018. Cancer Res 2018, 78: Abstract nr 976.
-
Robinson JL, Holmes KA, Carroll JS: FOXA1 mutations in hormone-dependent cancers. Front Oncol 2013, 3: 20.
-
Calo E, Wysocka J: Modification of enhancer chromatin: what, how, and why? Mol Cell 2013, 49(5): 825-837.
-
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al: Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013, 6(269): pl1.
-
Lv S, Ji L, Chen B, Liu S, Lei C, Liu X, Qi X, Wang Y, Lai-Han Leung E, Wang H et al: Histone methyltransferase KMT2D sustains prostate carcinogenesis and metastasis via epigenetically activating LIFR and KLF4. Oncogene 2018, 37(10): 1354-1368.
-
Dawkins JB, Wang J, Maniati E, Heward JA, Koniali L, Kocher HM, Martin SA, Chelala C, Balkwill FR, Fitzgibbon J et al: Reduced Expression of Histone Methyltransferases KMT2C and KMT2D Correlates with Improved Outcome in Pancreatic Ductal Adenocarcinoma. Cancer Res 2016, 76(16): 4861-4871.
-
Ford DJ, Dingwall AK: The cancer COMPASS: navigating the functions of MLL complexes in cancer. Cancer Genet 2015, 208(5): 178-191.
-
Jung SH, Shin S, Kim MS, Baek IP, Lee JY, Lee SH, Kim TM, Lee SH, Chung YJ: Genetic Progression of High Grade Prostatic Intraepithelial Neoplasia to Prostate Cancer. Eur Urol 2016, 69(5): 823-830.
-
Kantidakis T, Saponaro M, Mitter R, Horswell S, Kranz A, Boeing S, Aygun O, Kelly GP, Matthews N, Stewart A et al: Mutation of cancer driver MLL2 results in transcription stress and genome instability. Genes Dev 2016, 30(4): 408-420.
-
Kote-Jarai Z, Leongamornlert D, Saunders E, Tymrakiewicz M, Castro E, Mahmud N, Guy M, Edwards S, O'Brien L, Sawyer E et al: BRCA2 is a moderate penetrance gene contributing to young-onset prostate cancer: implications for genetic testing in prostate cancer patients. Br J Cancer 2011, 105(8): 1230-1234.
-
Taylor RA, Fraser M, Rebello RJ, Boutros PC, Murphy DG, Bristow RG, Risbridger GP: The influence of BRCA2 mutation on localized prostate cancer. Nat Rev Urol 2019, 16(5): 281-290.
-
Taylor RA, Fraser M, Livingstone J, Espiritu SM, Thorne H, Huang V, Lo W, Shiah YJ, Yamaguchi TN, Sliwinski A et al: Germline BRCA2 mutations drive prostate cancers with distinct evolutionary trajectories. Nat Commun 2017, 8: 13671.
-
Edwards SM, Evans DG, Hope Q, Norman AR, Barbachano Y, Bullock S, Kote-Jarai Z, Meitz J, Falconer A, Osin P et al: Prostate cancer in BRCA2 germline mutation carriers is associated with poorer prognosis. Br J Cancer 2010, 103(6): 918-924.
-
Watanabe M, Kakiuchi H, Kato H, Shiraishi T, Yatani R, Sugimura T, Nagao M: APC gene mutations in human prostate cancer. Jpn J Clin Oncol 1996, 26(2): 77-81.
-
Khemlina G, Ikeda S, Kurzrock R: Molecular landscape of prostate cancer: implications for current clinical trials. Cancer Treat Rev 2015, 41(9): 761-766.
-
Bruxvoort KJ, Charbonneau HM, Giambernardi TA, Goolsby JC, Qian CN, Zylstra CR, Robinson DR, Roy-Burman P, Shaw AK, Buckner-Berghuis BD et al: Inactivation of Apc in the mouse prostate causes prostate carcinoma. Cancer Res 2007, 67(6): 2490-2496.
-
Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, Shmoish M, Nativ N, Bahir I, Doniger T, Krug H et al: GeneCards Version 3: the human gene integrator. Database (Oxford) 2010, 2010: 1-16.
-
Chen Y, Li J, Yu X, Li S, Zhang X, Mo Z, Hu Y: APC gene hypermethylation and prostate cancer: a systematic review and meta-analysis. Eur J Hum Genet 2013, 21(9): 929-935.
-
Brewster SF, Browne S, Brown KW: Somatic allelic loss at the DCC, APC, nm23-H1 and p53 tumor suppressor gene loci in human prostatic carcinoma. J Urol 1994, 151(4): 1073-1077.
-
Yegnasubramanian S, Kowalski J, Gonzalgo ML, Zahurak M, Piantadosi S, Walsh PC, Bova GS, De Marzo AM, Isaacs WB, Nelson WG: Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res 2004, 64(6): 1975-1986.
Annals of urologic oncology
p-ISSN: 2617-7765, e-ISSN: 2617-7773
 Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.
Copyright © Ann Urol Oncol. This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivatives 4.0 International (CC BY-NC-ND 4.0) License.